- Patient Care & Health Information
- Diseases & Conditions
- Cystic fibrosis
- Cystic fibrosis FAQs
Pulmonologist Sarah Chalmers, M.D., answers the most frequently asked questions about cystic fibrosis.
Hello. I'm Dr. Sarah Chalmers, a pulmonologist at Mayo Clinic. And I'm here to answer some of the important questions you may have about cystic fibrosis.
Just because your baby's newborn screen came back positive does not mean that your baby has cystic fibrosis. Most babies who have a positive screening actually don't have CF. The newborn screen looks at a substance in the blood that is elevated in cystic fibrosis, but it can be elevated in other conditions as well, even premature birth. Some states also test for a gene mutation, but even if this comes back positive, it doesn't mean your baby has the disease. People with only one mutation are called carriers. It's very common in the United States and one in 20 people are CF gene mutation carriers. If your baby has a positive cystic fibrosis screen, they will need to see their doctor and have a sweat chloride test to see if they do have cystic fibrosis.
CF gene mutations are actually passed from parent to children in a specific pattern called autosomal recessive. Each parent passes one CF gene to their child, and therefore each person has two CF genes. To get the disease, both genes have to have a mutation. People with one CF gene are called carriers. If a parent is a carrier, there's a 50 percent chance they'll pass on the gene with a mutation to their child. If both parents pass on a normal gene, or only one parent passes a gene with a mutation, the child will not have CF. If both parents pass on a gene with a mutation, then the baby will have two genes with the mutation and will likely get the disease. If both parents are CF mutation carriers, there's a 25 percent chance that each one of their babies will be born with cystic fibrosis.
So both males and females can get cystic fibrosis. But females tend to have more symptoms, more lung infections, and they tend to start these symptoms of infections earlier in life as compared to males. No one knows for sure why this is so.
Actually, nearly 10 percent of cases of CF are diagnosed in adulthood. You're born with cystic fibrosis, but there are several reasons why it may not be diagnosed during childhood. Prior to 2010, some states didn't even screen for cystic fibrosis. So if you were born before 2010, you may not have received a newborn screening test for cystic fibrosis as a baby. Some gene mutations cause very mild disease and symptoms may go unnoticed until adulthood.
CF symptoms, how the disease affects the patient's organs and how it impacts their life is very different from one person to the next. Some people have very mild disease with only one organ affected and very few symptoms, while others have more severe disease with troublesome symptoms and multiple organs that are affected. Many factors including gene mutation type determine the impact on the patient. But your cystic fibrosis care team can work with you as an individual patient to create a personalized treatment plan that meets your individual needs.
Fertility is affected in both men and women with cystic fibrosis. Women with CF have thicker cervical mucus and they may also have irregular menstrual cycles. So it may take longer for women with CF to become pregnant. But most can become pregnant, have a normal pregnancy and a normal delivery. Almost all men with CF have infertility. Men with CF make normal sperm, but the sperm canal is absent. Because they still make sperm, assisted reproductive technologies can be used to help male CF patients have biologic children. Whether your children get CF or not depends on the combination of genes passed on from you and your significant other and can range from zero chance if neither parent has a gene mutation to a near 100% chance if both parents have CF.
Always be honest with your health care team. Let us know which medications you're taking and how often you're doing your treatments. Write down your questions before you come to your appointment so that we can make sure that we are meeting your needs. Thanks for your time. And we wish you well.
To diagnose cystic fibrosis, doctors typically do a physical exam, review your symptoms and conduct several tests.

Newborn screening and diagnosis
Every state in the U.S. now routinely screens newborns for cystic fibrosis. Early diagnosis means that treatment can begin immediately.
In one screening test, a blood sample is checked for higher than normal levels of a chemical called immunoreactive trypsinogen (IRT), which is released by the pancreas. A newborn's IRT levels may be high because of premature birth or a stressful delivery. For that reason, other tests may be needed to confirm a diagnosis of cystic fibrosis.
To evaluate if an infant has cystic fibrosis, doctors may also conduct a sweat test once the infant is at least 2 weeks old. A sweat-producing chemical is applied to a small area of skin. Then the sweat is collected to test it and see if it's saltier than normal. Testing done at a care center accredited by the Cystic Fibrosis Foundation helps ensure reliable results.
Doctors may also recommend genetic tests for specific defects on the gene responsible for cystic fibrosis. Genetic tests may be used in addition to checking the IRT levels to confirm the diagnosis.
Testing of older children and adults
Cystic fibrosis tests may be recommended for older children and adults who weren't screened at birth. Your doctor may suggest genetic and sweat tests for CF if you have recurring bouts of an inflamed pancreas, nasal polyps, chronic sinus or lung infections, bronchiectasis, or male infertility.
- Care at Mayo Clinic
Our caring team of Mayo Clinic experts can help you with your cystic fibrosis-related health concerns Start Here
More Information
Cystic fibrosis care at Mayo Clinic
- Genetic testing
There is no cure for cystic fibrosis, but treatment can ease symptoms, reduce complications and improve quality of life. Close monitoring and early, aggressive intervention is recommended to slow the progression of CF , which can lead to a longer life.
Managing cystic fibrosis is complex, so consider getting treatment at a center with a multispecialty team of doctors and medical professionals trained in CF to evaluate and treat your condition.
The goals of treatment include:
- Preventing and controlling infections that occur in the lungs
- Removing and loosening mucus from the lungs
- Treating and preventing intestinal blockage
- Providing adequate nutrition
Medications
Options include:
- Medications that target gene mutations, including a new medication that combines three drugs to treat the most common genetic mutation causing CF and is considered a major achievement in treatment
- Antibiotics to treat and prevent lung infections
- Anti-inflammatory medications to lessen swelling in the airways in your lungs
- Mucus-thinning drugs, such as hypertonic saline, to help you cough up the mucus, which can improve lung function
- Inhaled medications called bronchodilators that can help keep your airways open by relaxing the muscles around your bronchial tubes
- Oral pancreatic enzymes to help your digestive tract absorb nutrients
- Stool softeners to prevent constipation or bowel obstruction
- Acid-reducing medications to help pancreatic enzymes work better
- Specific drugs for diabetes or liver disease, when appropriate
Medications that target genes
For those with cystic fibrosis who have certain gene mutations, doctors may recommend cystic fibrosis transmembrane conductance regulator (CFTR) modulators. These newer medications help improve the function of the faulty CFTR protein. They may improve lung function and weight, and reduce the amount of salt in sweat.
The FDA has approved these medications for treating CF in people with one or more mutations in the CFTR gene:
- The newest combination medication containing elexacaftor, ivacaftor and tezacaftor (Trikafta) is approved for people age 12 years and older and considered a breakthrough by many experts.
- The combination medication containing tezacaftor and ivacaftor (Symdeko) is approved for people age 6 years and older.
- The combination medication containing lumacaftor and ivacaftor (Orkambi) is approved for people who are age 2 years and older.
- Ivacaftor (Kalydeco) has been approved for people who are 6 months and older.
A milestone treatment for CF
Tim Myer has lived with cystic fibrosis (CF) his whole life. He was awaiting a lung transplant when a new medication approved by the Food and Drug Administration changed everything. Myer and Dr. Mark Wylam, a Mayo Clinic pulmonologist who is Myer's physician, share the remarkable story.
Doctors may conduct liver function tests and eye exams before prescribing these medications. While taking these drugs, testing on a regular basis is needed to check for side effects such as liver function abnormalities and cataracts. Ask your doctor and pharmacist for information on possible side effects and what to watch for.
Keep regular follow-up appointments so your doctor can monitor you while taking these medications. Talk to your doctor about any side effects that you experience.
Airway clearance techniques

Using a personalized approach, a Mayo Clinic respiratory therapist discusses inflatable vest therapy with an adult who has cystic fibrosis.
Airway clearance techniques — also called chest physical therapy (CPT) — can relieve mucus obstruction and help to reduce infection and inflammation in the airways. These techniques loosen the thick mucus in the lungs, making it easier to cough up.
Airway clearing techniques are usually done several times a day. Different types of CPT can be used to loosen and remove mucus, and a combination of techniques may be recommended.
- A common technique is clapping with cupped hands on the front and back of the chest.
- Certain breathing and coughing techniques also may be used to help loosen the mucus.
- Mechanical devices can help loosen lung mucus. Devices include a tube that you blow into and a machine that pulses air into the lungs (vibrating vest). Vigorous exercise also may be used to clear mucus.
Your doctor will instruct you on the type and frequency of chest physical therapy that's best for you.
Pulmonary rehabilitation
Your doctor may recommend a long-term program that may improve your lung function and overall well-being. Pulmonary rehabilitation is usually done on an outpatient basis and may include:
- Physical exercise that may improve your condition
- Breathing techniques that may help loosen mucus and improve breathing
- Nutritional counseling
- Counseling and support
- Education about your condition
Surgical and other procedures
Options for certain conditions caused by cystic fibrosis include:
- Nasal and sinus surgery. Your doctor may recommend surgery to remove nasal polyps that obstruct breathing. Sinus surgery may be done to treat recurrent or chronic sinusitis.
- Oxygen therapy. If your blood oxygen level declines, your doctor may recommend that you breathe pure oxygen to prevent high blood pressure in the lungs (pulmonary hypertension).
- Noninvasive ventilation. Typically used while sleeping, noninvasive ventilation uses a nose or mouth mask to provide positive pressure in the airway and lungs when you breathe in. It's often used in combination with oxygen therapy. Noninvasive ventilation can increase air exchange in the lungs and decrease the work of breathing. The treatment may also help with airway clearance.
- Feeding tube. Cystic fibrosis interferes with digestion, so you can't absorb nutrients from food very well. Your doctor may suggest using a feeding tube to deliver extra nutrition. This tube may be a temporary tube inserted into your nose and guided to your stomach, or the tube may be surgically implanted in the abdomen. The tube can be used to give extra calories during the day or night and does not prevent eating by mouth.
- Bowel surgery. If a blockage develops in your bowel, you may need surgery to remove it. Intussusception, where a segment of intestine has telescoped inside an adjacent section of intestine, also may require surgical repair.
Lung transplant. If you have severe breathing problems, life-threatening lung complications or increasing resistance to antibiotics for lung infections, lung transplantation may be an option. Because bacteria line the airways in diseases that cause permanent widening of the large airways (bronchiectasis), such as cystic fibrosis, both lungs need to be replaced.
Cystic fibrosis does not recur in transplanted lungs. However, other complications associated with CF — such as sinus infections, diabetes, pancreas conditions and osteoporosis — can still occur after a lung transplant.
- Liver transplant. For severe cystic fibrosis-related liver disease, such as cirrhosis, liver transplant may be an option. In some people, a liver transplant may be combined with lung or pancreas transplants.
- Home enteral nutrition
- Lung transplant
Clinical trials
Explore Mayo Clinic studies testing new treatments, interventions and tests as a means to prevent, detect, treat or manage this condition.
Lifestyle and home remedies
You can manage your condition and minimize complications in several ways.
Pay attention to nutrition and fluid intake
Cystic fibrosis can cause malnourishment because the enzymes needed for digestion can't reach your small intestine, preventing food from being absorbed. People with CF may need a much higher number of calories daily than do people without the condition.
A healthy diet is important to growth and development and to maintain good lung function. It's also important to drink lots of fluids, which can help thin the mucus in your lungs. You may work with a dietitian to develop a nutrition plan.
Your doctor may recommend:
- Pancreatic enzyme capsules with every meal and snack
- Medications to suppress acid production
- Supplemental high-calorie nutrition
- Special fat-soluble vitamins
- Extra fiber to prevent intestinal blockage
- Extra salt, especially during hot weather or before exercising
- Adequate water intake, especially during hot weather
Keep vaccinations up to date
In addition to receiving other usual childhood vaccines, people with cystic fibrosis should have the annual flu vaccine and any other vaccines their doctors recommend, such as the vaccine to prevent pneumonia. CF doesn't affect the immune system, but children with CF are more likely to develop complications when they become sick.
Regular exercise helps loosen mucus in your airways and strengthens your heart. Because people with cystic fibrosis are living longer, maintaining good cardiovascular fitness for a healthy life is important. Anything that gets you moving, including walking and biking, can help.
Eliminate smoke
Don't smoke, and don't allow other people to smoke around you or your child. Secondhand smoke is harmful for everyone, but especially for people with cystic fibrosis, as is air pollution.
Encourage hand-washing
Teach all the members of your family to wash their hands thoroughly before eating, after using the bathroom, when coming home from work or school, and after being around a person who is sick. Hand-washing is the best way to protect against infection.
Attend medical appointments
You'll have ongoing care from your doctor and other medical professionals.
- Make sure to attend your regular follow-up appointments.
- Take your medications as prescribed and follow therapies as instructed.
- Talk to your doctor about how to manage symptoms and the warning signs of serious complications.
Coping and support
If you or someone you love has cystic fibrosis, you may experience strong emotions such as depression, anxiety, anger or fear. These issues may be especially common in teens. These tips may help.
- Find support. Talking openly about how you feel can help. It also may help to talk with others who are dealing with the same issues. That might mean joining a support group for yourself, or finding a support group for parents of children with cystic fibrosis. Older children with CF may want to join a CF group to meet and talk with others who have the disorder.
- Seek professional help. If you or your child is depressed or anxious, it may help to meet with a mental health professional. He or she can talk with you about feelings and coping strategies, and may suggest medications or other treatments as well.
- Spend time with friends and family. Having their support can help you manage stress and reduce anxiety. Ask your friends or family for help if you need it.
- Take time to learn about cystic fibrosis. If your child has cystic fibrosis, encourage him or her to learn about CF . Find out how medical care is managed for children with CF as they grow older and reach adulthood. Talk with your doctor if you have questions about care.
Preparing for your appointment
Make an appointment with your doctor if you or your child has signs or symptoms common to cystic fibrosis. After the initial evaluation, you may be referred to a doctor trained in evaluating and treating CF .
Here's some information to help you prepare for your appointment, as well as what to expect from your doctor.
What you can do
You may want to prepare answers to these questions:
- What symptoms are you or your child experiencing?
- When did the symptoms start?
- Does anything make the symptoms better or worse?
- Has anyone in your family ever had cystic fibrosis?
- Has growth been normal and weight been stable?
What to expect from your doctor
After getting detailed information about the symptoms and your family's medical history, your doctor may order tests to help with diagnosis and plan treatment.
Living with cystic fibrosis?
Connect with others like you for support and answers to your questions in the Transplants support group on Mayo Clinic Connect, a patient community.
Transplants Discussions

336 Replies Sat, Jul 20, 2024

21 Replies Mon, Jul 15, 2024

26 Replies Thu, Jul 11, 2024
- Symdeko (prescribing information). Vertex Pharmaceuticals Inc.; 2019. https://www.symdeko.com/how-symdeko-works. Accessed July 1, 2019.
- Kalydeco (prescribing information). Vertex Pharmaceuticals Inc.; 2019. https://www.kalydeco.com/. Accessed July 1, 2019.
- Orkambi (prescribing information). Vertex Pharmaceuticals Inc.; 2018. https://www.orkambi.com/. Accessed July 1, 2019.
- Chest physiotherapy compared to no chest physiotherapy for cystic fibrosis. Cochrane Database of Systematic Reviews. 2015; doi:10.1002/14651858.CD001401.pub3.
- Cystic fibrosis. National Heart, Lung, and Blood Institute. https://www.nhlbi.nih.gov/health-topics/cystic-fibrosis. Accessed July 1, 2019.
- Cystic fibrosis. Genetics Home Reference. https://ghr.nlm.nih.gov/condition/cystic-fibrosis. Accessed July 1, 2019.
- AskMayoExpert. Cystic fibrosis. Mayo Clinic; 2017.
- Bronchiectasis. National Heart, Lung, and Blood Institute. https://www.nhlbi.nih.gov/health-topics/bronchiectasis. Accessed July 1, 2019.
- Rafeeq MM, et al. Cystic fibrosis: Current therapeutic targets and future approaches. Journal of Translational Medicine. 2017; doi:10.1186/s12967-017-1193-9.
- Cystic fibrosis. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pediatrics/cystic-fibrosis-cf/cystic-fibrosis. Accessed July 1, 2019.
- Frequently asked questions: Pregnancy FAQ171: Cystic fibrosis: Prenatal screening and diagnosis. American College of Obstetricians and Gynecologists. https://www.acog.org/Patients/FAQs/Cystic-Fibrosis-Prenatal-Screening-and-Diagnosis?IsMobileSet=false. Accessed July 1, 2019.
- Simon RH. Cystic fibrosis: Treatment with CFTR modulators. https://www.uptodate.com/contents/search. Accessed July 1, 2019.
- Simon RH. Cystic fibrosis: Overview of treatment of lung disease. https://www.uptodate.com/contents/search. Accessed July 1, 2019.
- Solomon M, et al. Nutritional issues in cystic fibrosis. Clinics in Chest Medicine. 2016; doi:10.1016/j.ccm.2015.11.009.
- Savant AP, et al. Cystic fibrosis year in review 2018, part 1. Pediatric Pulmonology. 2019; doi:10.1002/ppul.24361.
- Savant AP, et al. Cystic fibrosis year in review 2018, part 2. Pediatric Pulmonology. 2019; doi:10.1002/ppul.24365.
- Brown A. Allscripts EPSi. Mayo Clinic. June 14, 2019.
- Drug trials snapshots: Trikafta. U.S. Food and Drug Administration. https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trials-snapshots-trikafta. Accessed Dec. 21, 2019.
- Trikafta (prescribing information). Vertex Pharmaceuticals Inc.; 2019. https://www.trikaftahcp.com/. Accessed Nov. 5, 2019.
- Boesch RP (expert opinion). Mayo Clinic. Dec. 11, 2019.
- Kayani K, et al. Cystic fibrosis-related diabetes. Frontiers in Endocrinology. 2018; doi:10.3389/fendo.2018.00020.
- van de Peppel IP, et al. Diagnosis, follow-up and treatment of cystic fibrosis-related liver disease. Current Opinion in Pulmonary Medicine. 2017; doi:10.1097/MCP.0000000000000428.
- Care centers. Cystic Fibrosis Foundation. https://www.cff.org/Care/Care-Centers/. Accessed Nov. 20, 2019.
- Moran F, et al. Non-invasive ventilation for cystic fibrosis. Cochrane Database of Systematic Reviews. 2017; doi:10.1002/14651858.CD002769.pub5.
- What is cystic fibrosis? A Mayo Clinic expert explains
Associated Procedures
Products & services.
- A Book: Mayo Clinic Family Health Book
- Newsletter: Mayo Clinic Health Letter — Digital Edition
Mayo Clinic in Rochester, Minnesota, Mayo Clinic in Phoenix/Scottsdale, Arizona, and Mayo Clinic in Jacksonville, Florida, have been recognized among the top Pulmonology hospitals in the nation for 2024-2025 by U.S. News & World Report.
- Symptoms & causes
- Diagnosis & treatment
- Doctors & departments
Mayo Clinic does not endorse companies or products. Advertising revenue supports our not-for-profit mission.
- Opportunities
Mayo Clinic Press
Check out these best-sellers and special offers on books and newsletters from Mayo Clinic Press .
- Mayo Clinic on Incontinence - Mayo Clinic Press Mayo Clinic on Incontinence
- The Essential Diabetes Book - Mayo Clinic Press The Essential Diabetes Book
- Mayo Clinic on Hearing and Balance - Mayo Clinic Press Mayo Clinic on Hearing and Balance
- FREE Mayo Clinic Diet Assessment - Mayo Clinic Press FREE Mayo Clinic Diet Assessment
- Mayo Clinic Health Letter - FREE book - Mayo Clinic Press Mayo Clinic Health Letter - FREE book
Double your impact on fighting cancer
Make a gift before July 31 and it can go twice as far to fight cancer.
Masks Strongly Recommended but Not Required in Maryland, Starting Immediately
Due to the downward trend in respiratory viruses in Maryland, masking is no longer required but remains strongly recommended in Johns Hopkins Medicine clinical locations in Maryland. Read more .
- Vaccines
- Masking Guidelines
- Visitor Guidelines
- Cystic Fibrosis
What is cystic fibrosis?
Cystic fibrosis (CF) is an inherited life-threatening disease that affects many organs. It causes changes in the electrolyte transport system. People with CF have problems with the glands that make sweat and mucus. CF makes mucus thicker. Symptoms start in childhood. On average, people with CF live into their mid to late 30s. But new treatments are increasing life expectancy.
CF affects several organ systems, including:
Respiratory system
Digestive system
Reproductive system
Some people carry the CF gene without being affected by the disease. They often don't know that they are carriers.
How does CF affect the respiratory system?
With CF, there is an abnormal electrolyte transport system. The normal thin secretions in the lungs become very thick and hard to move. These thick secretions raise the risk for frequent respiratory infections.
Respiratory infections that keep coming back lead to more damage in the lungs. Over time, this causes permanent loss of lung function.
Because of the high rate of infection in the lower respiratory tract, people with CF may develop a chronic cough and blood in the sputum. The cough is often worse in the morning or after activity. They can develop lung collapse (pneumothorax).
People with CF also have upper respiratory tract symptoms. Some have nasal polyps that need surgery to be removed. Nasal polyps are small bumps of tissue from the lining of the nose. They can block and irritate the nasal cavity. People with CF also have higher rates of sinus infections.
How does cystic fibrosis affect the digestive system?
CF mainly affects the pancreas. The pancreas secretes substances that aid digestion and help control blood sugar levels.
The secretions from the pancreas also become thick and can clog the ducts of the pancreas. This may cause a decrease in the secretion of enzymes from the pancreas that normally help digest food. A person with CF has trouble absorbing proteins, fats, and vitamins A, D, E, and K.
The problems with the pancreas can get so severe that some of the cells in the pancreas die. Over time, this may lead to glucose intolerance. It may also lead to cystic fibrosis-related diabetes (CFRD). This is a unique type of insulin-dependent diabetes.
Some CF symptoms may be from its effect on the digestive tract. These include:
Bulky, greasy stools
The lower end of the bowel comes out of the anus (rectal prolapse)
Delayed puberty
Fat in the stools
Stomach pain
Bloody diarrhea
The liver may also be affected. A small number of people may develop liver disease. Symptoms of liver disease include:
Enlarged liver
Swollen belly
Yellow color to the skin (jaundice)
Vomiting blood
How does CF affect the reproductive system?
Most males with CF have blockage of the sperm canal. This is called congenital bilateral absence of the vas deferens (CBAVD). This results from the thick secretions clogging the vas deferens and keeping them from developing correctly. It causes infertility because sperm can't travel out of the body. There are some newer methods that allow men with CF to have children. Discuss these with your healthcare provider. Women with CF have an increase in thick cervical mucus that may lead to a decrease in fertility. They may also have irregular ovulation. But many women with CF are able to have children.
What causes cystic fibrosis?
CF is a genetic disease. This means that CF is inherited.
Changes (mutations) in a gene called the CFTR (cystic fibrosis transmembrane conductance regulator) gene cause CF. The CFTR mutations cause changes in the body’s electrolyte transport system. Electrolytes are substances in blood that are vital to cell function. The main result of these transport system changes is seen in the body secretions, such as mucus and sweat.
The CFTR gene is large and complex. There are many different mutations in this gene that have been linked to CF.
A person will be born with CF only if 2 CF genes are inherited: 1 from the mother and 1 from the father.
Who is at risk for cystic fibrosis?
Cystic fibrosis is inherited. A person with CF had both parents pass the changed gene to them. The birth of a child with CF is often a total surprise to a family. Most of the time there is no family history of CF. Caucasian people are more likely to have CF than people of African, Asian, or Hispanic ancestry.
What are the symptoms of cystic fibrosis?
Symptoms can be different for each person. The severity of symptoms can vary, too. Symptoms may include:
Thick mucus that clogs certain organs such as the lungs, pancreas, and intestines. This may cause malnutrition, poor growth, frequent respiratory infections, breathing problems, and ongoing (chronic) lung disease.
Many other health problems can point to cystic fibrosis, as well. These include:
Nasal polyps
Clubbed fingers and toes. This means thickened fingertips and toes because of less oxygen in the blood.
Collapsed lung, often due to intense coughing
Coughing up blood
Enlargement of the right side of the heart due to increased pressure in the lungs (Cor pulmonale)
Too much gas in the intestines
Liver disease
Inflammation of the pancreas (pancreatitis) that causes severe pain in the belly
Congenital bilateral absence of the vas deferens (CBAVD) in males. This causes blockages of the sperm canal.
Babies born with CF often show symptoms by age 2. But some children may not show symptoms until later in life. The following symptoms may mean a child has CF. Babies with these signs may have more testing for CF:
Diarrhea that does not go away
Bad-smelling stools
Greasy stools
Frequent wheezing
Frequent pneumonia or other lung infections
Persistent cough
Skin that tastes like salt
Poor growth despite having a good appetite
The symptoms of CF may seem like other conditions or health problems. See a healthcare provider for a diagnosis.
How is cystic fibrosis diagnosed?
All U.S. states require that newborns be tested for CF. This means that parents can know if their baby has the disease. They can take precautions and watch for early signs of problems. Most cases of cystic fibrosis are found during newborn screening. Babies will have a full health history and physical exam.
Tests for CF include a sweat test to measure the amount of salt (sodium chloride) present. This test may be used if a person has symptoms of CF or if a newborn screening suggests that a baby may have CF. Higher than normal amounts of sodium chloride suggest CF. Other tests depend on which body system is affected. These tests may include:
Chest X-rays, ultrasound, and CT scans
Blood tests
Lung function tests
Sputum cultures
Stool tests
For babies who don't make enough sweat, blood tests may be used.
How is cystic fibrosis treated?
There is currently no cure for CF. Scientists are investigating gene therapy. Some patients with advanced disease may be considered for surgeries like lung and pancreas transplant.
Goals of treatment are to ease symptoms, prevent and treat complications, and slow the progress of the disease.
Treatment generally focuses on the following 2 areas.
Managing lung problems
This may include:
Physical therapy
Airway clearance therapy, including chest physical therapy, to loosen and clear mucus
Medicines to thin mucus and help breathing
Antibiotics to treat infections
Anti-inflammatory medicines
Lung transplant may be a choice for people with end-stage lung disease. The type of transplant done is often a heart-lung transplant or a double lung transplant. Not everyone is a candidate for a lung transplant. Discuss this option with your healthcare provider.
Managing digestive problems
A healthy diet that's high in calories
Pancreatic enzymes to aid digestion
Vitamin supplements
Treatments for intestinal blockages
What are possible complications of cystic fibrosis?
CF has serious complications, including:
Worsening lung function, leading to the inability to do daily activities
Lung infections
Lung collapse (pneumothorax)
Inflammation of the pancreas
Cirrhosis (liver disease)
Vitamin deficiencies
Inability for a child to grow and develop (failure to thrive)
Infertility
Cystic fibrosis-related diabetes (CFRD)
Gastroesophageal reflux disease (GERD). With this disease, stomach contents rise up into the esophagus and can cause serious damage.
Can cystic fibrosis be prevented?
Cystic fibrosis is caused by an inherited gene change (mutation). Testing for the CF gene is recommended for anyone who has a family member with the disease. It is also advised for someone whose partner is a known carrier of CF or affected with CF.
Testing for the CF gene can be done from a small blood sample. Or it can be done from a cheek swab. This is a brush rubbed against the inside of your cheek to get cells for testing. Labs generally test for the most common CF gene mutations.
There are many people with CF whose mutations have not been identified. Experts have not discovered all the genetic errors that cause CF. This means that a person can still be a CF carrier even if no mutations were found by testing. There are limits to CF testing.
Two people who are carriers of the CF gene have a 1 in 4 chance of having a child with CF. If both partners have the CF gene and are thinking about having a child, they have some choices:
Choose prenatal diagnosis. This means the baby can be checked for CF between 10 to 13 weeks and 15 to 20 weeks during pregnancy.
End a pregnancy.
Prepare to have your child with CF. Talk to healthcare providers and parents of children with CF.
Prepare to establish a treatment plan for your child with CF. Talk to healthcare providers about what your newborn's needs may be.
Don't become pregnant.
Explore surrogacy, adoption, or other ways to start a family.
Living with cystic fibrosis
If you have been diagnosed with CF, here are some ways to help manage it:
It's important to stay up-to-date with vaccines. They reduce the risk of infection. Ask your healthcare provider what vaccines you need. This may include the influenza, COVID-19, and pneumococcal vaccines.
You may need to take inhaled antibiotics for the long term to prevent lung infections.
You may need medicines to help with digestion.
Your healthcare provider may advise vitamin and mineral supplements.
The physical, emotional, and financial stress that CF places on a family is enormous. Ask your healthcare provider for resources to help support your family and manage the disease. Online and in-person family support groups and peer support groups for the person with CF can also be very helpful.
Key points about cystic fibrosis
Cystic fibrosis (CF) is an inherited life-threatening disease that affects many organs. It causes changes in the electrolyte transport system.
People with CF have problems in the glands that produce sweat and mucus.
CF causes thick mucus that clogs certain organs such as the lungs, pancreas, and intestines. This may cause malnutrition, poor growth, frequent respiratory infections, breathing problems, and chronic lung disease.
All U.S. states require that newborns be tested for CF. This is how most cases are diagnosed.
There is no cure for CF. Goals of treatment are to ease symptoms, prevent and treat complications, and slow the progress of the disease.
Tips to help you get the most from a visit to your healthcare provider:
Know the reason for your visit and what you want to happen.
Before your visit, write down questions you want answered.
Bring someone with you to help you ask questions and remember what your provider tells you.
At the visit, write down the name of a new diagnosis and any new medicines, treatments, or tests. Also write down any new instructions your provider gives you.
Know why a new medicine or treatment is prescribed and how it will help you. Also know what the side effects are.
Ask if your condition can be treated in other ways.
Know why a test or procedure is advised and what the results could mean.
Know what to expect if you do not take the medicine or have the test or procedure.
If you have a follow-up appointment, write down the date, time, and purpose for that visit.
Know how you can contact your provider if you have questions. Ask how to contact your healthcare team on weekends, holidays, and evenings in case you have urgent concerns.
Find a Doctor
Specializing In:
Cystic Fibrosis Liver Disease
Find a Treatment Center
- Pediatric Cystic Fibrosis (Johns Hopkins Children's Center)
Find Additional Treatment Centers at:
- Howard County Medical Center
- Sibley Memorial Hospital
- Suburban Hospital

Request an Appointment

Cystic Fibrosis: Sam's Story
- Help & Support
- Lung Health & Diseases
- Lung Disease Lookup
- Cystic Fibrosis (CF)
Cystic Fibrosis Symptoms and Diagnosis
What are the symptoms of cystic fibrosis.
Symptoms of lung disease can start in infancy, especially following upper respiratory viral infections. People with CF experience a small but progressive (worsening) loss in lung function with every passing year, leading to increased symptoms as you age. Some children remain relatively healthy throughout childhood and only start to experience a decline in their lung function when they are teenagers.
There is a wide range of severity in CF symptoms. Even within the same family, siblings can have different disease severity. Symptoms of CF can be classified into two main categories: respiratory and digestive.
The most common symptoms of CF respiratory tract disease are:
- Chronic coughing (dry or coughing up mucus)
- Recurring chest colds
- Wheezing or shortness of breath
- Frequent sinus infections
- Very salty-tasting skin
Digestive symptoms may include greasy, foul-smelling bowel movements, severe constipation or intestinal blockage and the inability to gain weight while being constantly hungry.
How Cystic Fibrosis Is Diagnosed
- Newborn screening . In the last decade, newborn screening has become standard and is now available in all 50 U.S. states. The newborn screen shows infants who have a high level of an enzyme called immunoreactive trypsin in their blood. This occurs when there is injury to the pancreas. The test is repeated if it is abnormal. Some states also combine this with testing for the most common gene mutation called deltaF508. The next step is to refer the infant for further testing as there are many “false-positive” tests. This entails taking a blood sample to check whether the infant has two genes that cause CF and/or performing a sweat test.
- Genetic testing . More than 2,000 different mutations of the CF gene have been identified. Most of them are quite rare, but a few are common, like the deltaF508 mutation that is found in at least 70% of individuals with CF. Genetic testing can determine the exact mutation in most cases. For couples who want to have children, genetic testing is also important as more than 10 million Americans are carriers of a CF gene. For every pregnancy, there is a one-in-four chance that the child will have CF when both parents are carriers.
- Sweat test . Sweat is collected from a small area on the child’s forearm, and the chloride levels are measured. Children with CF have high levels of chloride in their sweat because a lack of CFTR prevents the salt on the skin from being reabsorbed back into the sweat glands.
- Measuring nasal lining . Another way to confirm the diagnosis is to run a small electrical current across the nasal lining (epithelium). Different solutions are applied to the nasal lining and the electrical current is measured. People with CF respond very differently than those without CF to this test, and it may help confirm a diagnosis.
When to See Your Doctor
If you or a loved one have a family history of CF and have pulmonary symptoms, have been diagnosed with CF, or experience these symptoms, consult your healthcare provider and request an evaluation at an accredited CF center. The Cystic Fibrosis Foundation website has a tool to assist in locating a care center.
Reviewed and approved by the American Lung Association Scientific and Medical Editorial Review Panel.
Page last updated: June 7, 2024
A Breath of Fresh Air In Your Inbox
Join over 700,000 people who receive the latest news about lung health, including research, lung disease, air quality, quitting tobacco, inspiring stories and more!
Thanks for submitting your email.
Make a Donation
Your tax-deductible donation funds lung disease and lung cancer research, new treatments, lung health education, and more.
Become a Lung Health Insider
Thank you! You will now receive email updates from the American Lung Association.
Select Your Location
Select your location to view local American Lung Association events and news near you.
Change Language
Lung helpline.
Talk to our lung health experts at the American Lung Association. Our service is free and we are here to help you.
1-800-LUNG-USA
(1-800-586-4872)
Cystic Fibrosis
- Pathophysiology |
- Symptoms and Signs |
- Diagnosis |
- Treatment |
- Prognosis |
- Key Points |
- More Information |
Cystic fibrosis is an inherited disease of the exocrine glands affecting primarily the gastrointestinal and respiratory systems. It leads to chronic lung disease, exocrine pancreatic insufficiency, hepatobiliary disease, and abnormally high sweat electrolytes. Diagnosis is by sweat test or identification of 2 cystic fibrosis–causing gene variants in patients with a positive newborn screening test result or characteristic clinical features. Treatment is supportive through aggressive multidisciplinary care along with small-molecule correctors and potentiators targeting the cystic fibrosis transmembrane conductance regulator protein defect.
Cystic fibrosis (CF) is a life-threatening genetic disease, which in the United States occurs in about 1/3,300 White births, 1/15,300 Black births, and 1/32,000 Asian American births. There are approximately 40,000 people with CF living in the United States, and approximately 100,000 diagnosed with CF worldwide. Because of improved treatment and life expectancy, about 58% of patients in the United States with CF are now adults ( 1 ).
General reference
1. Cystic Fibrosis Foundation Patient Registry 2021 Annual Data Report Bethesda, Maryland 2022 Cystic Fibrosis Foundation. Accessed October 20, 2023.
Etiology of Cystic Fibrosis
Cystic fibrosis is carried as an autosomal recessive trait by about 3% of the White population. The responsible gene has been localized on the long arm of chromosome 7. It encodes a membrane-associated protein called the cystic fibrosis transmembrane conductance regulator (CFTR). The most common gene variant, F508del, occurs in about 85% of CF alleles; > 2000 less common CFTR variants have been identified.
CFTR is a cyclic adenosine monophosphate (cAMP)–regulated chloride channel, regulating chloride, sodium, and bicarbonate transport across epithelial membranes. A number of additional functions are considered likely. Disease manifests only in people who are homozygous. People who are heterozygous may show subtle abnormalities of epithelial electrolyte transport but are clinically unaffected.
The CFTR variants have been divided into 6 classes based on how the variant affects the function or processing of the CFTR protein. Patients with class I, II, or III variants are considered to have a more severe genotype that results in little or no CFTR function, whereas patients with 1 or 2 class IV, V, or VI variants are considered to have a milder genotype that results in residual CFTR function. However, there is no strict relationship between specific variants and disease manifestation, so clinical testing (ie, of organ function) rather than genotyping is a better guide to prognosis. CFTR variants can involve frameshift (a deletion or insertion in a DNA sequence that shifts the way a sequence is read) or nonsense (stop) mutations.
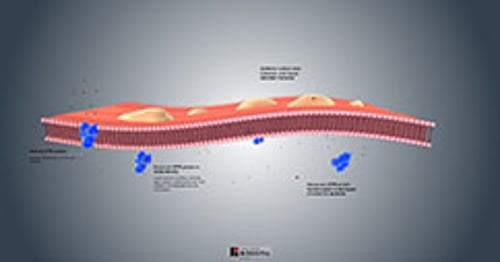
Pathophysiology of Cystic Fibrosis
Nearly all exocrine glands are affected in varying distribution and degree of severity. Glands may
Become obstructed by viscid mucus in the lumen (pancreas, intestinal glands, intrahepatic bile ducts, gallbladder, and submaxillary glands)
Appear histologically abnormal and produce excessive secretions (tracheobronchial and Brunner glands)
Appear histologically normal but secrete excessive sodium and chloride (sweat, parotid, and small salivary glands)
Respiratory
Although the lungs are generally histologically normal at birth, most patients develop signs of pulmonary disease beginning in infancy or early childhood. Mucus plugging and chronic bacterial infection, accompanied by a pronounced inflammatory response, damage the airways, ultimately leading to bronchiectasis and respiratory insufficiency. The course is characterized by episodic exacerbations with infection and progressive decline in pulmonary function.
Pulmonary damage is probably initiated by diffuse obstruction in the small airways by abnormally thick mucus secretions. Bronchiolitis and mucopurulent plugging of the airways occur secondary to obstruction and infection. Chronic inflammation secondary to the release of proteases and proinflammatory cytokines by cells in the airways also contributes to lung injury. Airway changes are more common than parenchymal changes, and emphysema is not prominent. About 50% of patients have bronchial hyperreactivity that may respond to bronchodilators.
In patients with advanced pulmonary disease, chronic hypoxemia results in muscular hypertrophy of the pulmonary arteries, pulmonary hypertension , and right ventricular hypertrophy.
The lungs of most patients are colonized by pathogenic bacteria. Early in the course, Staphylococcus aureus is the most common pathogen, but as the disease progresses, Pseudomonas aeruginosa , including multidrug-resistant strains, is frequently isolated. A mucoid variant of P. aeruginosa is uniquely associated with CF and results in a worse prognosis than nonmucoid P. aeruginosa .
In the United States, the prevalence of methicillin-resistant S. aureus ( MRSA ) in the respiratory tract is now about 25%; patients who are chronically infected with MRSA have more rapid decline in pulmonary function and lower survival rates than those who are not.
Colonization with Burkholderia cepacia complex occurs in about 2 to 3% of patients and may be associated with more rapid pulmonary deterioration.
Nontuberculous mycobacteria , including Mycobacterium avium complex and M. abscessus , are potential respiratory pathogens. Prevalence is around 14% and varies with age and geographic location. Differentiating infection from colonization can be challenging.
Other common respiratory pathogens include Stenotrophomonas maltophilia , Achromobacter xylosoxidans , and Aspergillus species.
Anaerobic bacteria and common respiratory viruses are frequently present in the respiratory tract of patients with CF, but their role in disease progression has not been well established.
Gastrointestinal
The pancreas, intestines, and hepatobiliary system are frequently affected. Exocrine pancreatic function is compromised in 85 to 95% of patients. An exception is a subset of patients who have certain CFTR variants with residual function, in whom pancreatic function is preserved. Patients with pancreatic insufficiency have malabsorption of fats, fat-soluble vitamins, and protein. Duodenal fluid is abnormally viscid and shows absence or diminution of enzyme activity and decreased bicarbonate concentration; stool trypsin and chymotrypsin are absent or diminished. Endocrine pancreatic dysfunction is less common, but impaired glucose tolerance or diabetes mellitus is present in about 2% of children, 20% of adolescents, and up to 50% of adults.
Bile duct involvement with bile stasis and biliary plugging leads to asymptomatic hepatic fibrosis in 30% of patients. About 3 to 4% of patients progress to irreversible multinodular biliary cirrhosis with varices and portal hypertension , usually by 12 years of age. Hepatocellular failure is a rare and late event. There is an increased incidence of cholelithiasis , which is usually asymptomatic.
Abnormally viscid intestinal secretions can cause meconium ileus in neonates and sometimes meconium plugging of the colon. Older children and adults also may have intermittent or chronic constipation and intestinal obstruction .
Other gastrointestinal (GI) problems include intussusception , volvulus, rectal prolapse , periappendiceal abscess, pancreatitis , an increased risk of cancer of the hepatobiliary tract and cancer of the GI tract (including of the pancreas ), gastroesophageal reflux , esophagitis, and an increased prevalence of Crohn disease and celiac disease .
Infertility occurs in 98% of adult men secondary to maldevelopment of the vas deferens or to other forms of obstructive azoospermia. In women, fertility is somewhat decreased secondary to viscid cervical secretions, although many women have carried pregnancies to term. Pregnancy outcome for both the mother and neonate is related to the mother's health.
Other complications include chronic rhinosinusitis , osteopenia/ osteoporosis , depression and anxiety, chronic pain, obstructive sleep apnea , other sleep disorders, renal stones , dialysis-dependent chronic kidney disease (possibly related to treatments as well as to CF), iron deficiency anemia , sensorineural hearing loss and tinnitus caused by exposure to ototoxic medications (especially aminoglycosides), and episodic arthralgias/arthritis.
Symptoms and Signs of Cystic Fibrosis
Fifty percent of patients not diagnosed through newborn screening present with pulmonary manifestations, often beginning in infancy. Recurrent or chronic infections manifested by cough, sputum production, and wheezing are common. Cough is the most common chronic symptom, often accompanied by sputum production, vomiting, and disturbed sleep. Intercostal retractions, use of accessory muscles of respiration, a barrel-chest deformity, digital clubbing, cyanosis, and a declining tolerance for exercise occur with disease progression. Upper respiratory tract involvement includes nasal polyposis and chronic or recurrent rhinosinusitis.
Pulmonary complications include pneumothorax , nontuberculous mycobacterial infection , hemoptysis , allergic bronchopulmonary aspergillosis (ABPA), and right heart failure secondary to pulmonary hypertension .
Meconium ileus due to obstruction of the ileum by viscid meconium may be the earliest sign and is present in about 10 to 20% of CF-affected neonates. It typically manifests with abdominal distention, vomiting, and failure to pass meconium. Some infants have intestinal perforation, with signs of peritonitis and shock. Infants with meconium plug syndrome have a delayed passage of meconium. They can have similar signs of obstruction or very mild and transient symptoms that go unnoticed. Older patients may have episodes of constipation or develop recurrent and sometimes chronic episodes of partial or complete small- or large-bowel obstruction (distal intestinal obstruction syndrome). Symptoms include crampy abdominal pain, change in stooling pattern, decreased appetite, and sometimes vomiting.
In infants without meconium ileus, disease onset may be heralded by a delay in regaining birth weight and inadequate weight gain at 4 to 6 weeks of age.
Occasionally, infants who are undernourished, especially if on hypoallergenic formula or soy formula, present with generalized edema secondary to protein malabsorption.
Pancreatic insufficiency is usually clinically apparent early in life and may be progressive. Manifestations include the frequent passage of bulky, foul-smelling, oily stools; abdominal protuberance; and poor growth pattern with decreased subcutaneous tissue and muscle mass despite a normal or voracious appetite. Clinical manifestations may occur secondary to deficiency of fat-soluble vitamins.
Rectal prolapse may occur in untreated infants and toddlers. Gastroesophageal reflux is relatively common among children and adults.
Excessive sweating in hot weather or with fever may lead to episodes of hyponatremic/hypochloremic dehydration and circulatory failure. In arid climates, infants may present with chronic metabolic alkalosis . Salt crystal formation and a salty taste on the skin are highly suggestive of CF.
Adolescents may have retarded growth and delayed onset of puberty.
Diagnosis of Cystic Fibrosis
Newborn screening.
May also be suggested by a positive prenatal screening test result, family history, or symptomatic presentation
Confirmed by a sweat test showing elevated sweat chloride on 2 occasions
Identifying 2 CF-causing variants (1 on each chromosome) is consistent with the diagnosis
May rarely be confirmed, in atypical cases, by demonstrating abnormal ion transport across the nasal epithelium or abnormal intestinal current measurements
Most cases of CF are first identified by newborn screening, but up to 10% are not diagnosed until adolescence or early adulthood. Despite advances in genetic testing, the sweat chloride test remains the standard for confirming a CF diagnosis in most cases because of its sensitivity and specificity, simplicity, and availability.
Universal newborn screening for CF is now standard in the United States. Screening is based on detecting an elevated concentration of immunoreactive trypsinogen (IRT) in the blood.
There are 2 methods of following up on an elevated IRT level. In one method, a second IRT is done, which, if also elevated, is followed by a sweat test. In the other, more commonly used method, an elevated IRT level is followed by CFTR mutation testing, and, if 1 or 2 variants are identified, then a sweat test is done. For diagnosis, both methods have 90 to 95% sensitivity.
Sweat testing
Normal: ≤ 30 mEq/L ( ≤ 30 mmol/L) (CF is unlikely.)
Intermediate: 30 to 59 mEq/L (30 to 59 mmol/L) (CF is possible.)
Abnormal: ≥ 60 mEq/L ( ≥ 60 mmol/L) (This result is consistent with CF.)
The results are valid after 48 hours of life, but an adequate sweat sample ( > 75 mg on filter paper or > 15 mcL in microbore tubing) may be difficult to obtain before 2 weeks of age. False-negative results are rare but may occur in the presence of edema and hypoproteinemia or an inadequate quantity of sweat. False-positive results are usually due to technical error. Transient elevation of sweat chloride concentration can result from psychosocial deprivation (eg, child abuse, neglect) and can occur in patients with anorexia nervosa. A positive sweat test result should be confirmed by a second sweat test or by identification of 2 CF-causing variants.
Intermediate sweat test results
A small subset of patients have a mild or partial CF phenotype and sweat chloride values that are persistently in the intermediate or even normal range. In addition, there are patients who have single-organ manifestations such as chronic or recurrent pancreatitis, isolated bronchiectasis, or congenital bilateral absence of the vas deferens along with findings suggestive of abnormal CFTR function. They do not meet criteria for a CF diagnosis and are classified as having a CFTR-related disorder. In some of these patients, the diagnosis of CF can be confirmed by the identification of 2 CF-causing variants, 1 on each chromosome. If 2 CF-causing variants are not identified, ancillary evaluations such as pancreatic function testing and pancreatic imaging, high-resolution chest CT, sinus CT, pulmonary function testing, urogenital evaluation in males, and bronchoalveolar lavage including assessment of microbial flora may be useful.
Additional potentially helpful diagnostic tests include expanded CFTR genetic analysis and measurement of nasal transepithelial potential difference (based on the observation of increased sodium reabsorption across epithelium that is relatively impermeable to chloride in patients with CF) and measurement of intestinal currents.
CFTR-related metabolic syndrome and CF screen positive, inconclusive diagnosis
Infants who have a positive newborn screening result and evidence of possible CFTR dysfunction but do not meet the diagnostic criteria for CF are classified as having CFTR-related metabolic syndrome (CRMS), also called CF screen positive, inconclusive diagnosis (CFSPID). CRMS/CFSPID is diagnosed in infants who have a positive newborn screen, are asymptomatic, and have either of the following:
Sweat chloride concentrations in the intermediate range and 0 or 1 CF-causing variant
Sweat chloride concentrations in the normal range and 2 CFTR variants, at least 1 of which has unclear phenotypic consequences
Most children with CRMS/CFSPID remain healthy, but over time around 10% will develop symptoms and meet criteria for a diagnosis of CF or a CF-related disorder. Patients with CRMS/CFSPID should be evaluated and monitored regularly in a CF care center.
Pancreatic tests
Pancreatic function should be assessed at the time of diagnosis, usually by measuring the concentration of human pancreatic elastase in stool. Human pancreatic elastase measurement is valid even in the presence of exogenous pancreatic enzymes. Infants who are initially pancreatic sufficient and who carry 2 "severe" variants should have serial measurements to detect progression to pancreatic insufficiency.
Respiratory assessment
Chest imaging is done at times of pulmonary deterioration or exacerbations and routinely every 1 to 2 years. High-resolution chest CT may be helpful to more precisely define the extent of lung damage and to detect subtle airway abnormalities. Chest x-rays and CT may show hyperinflation, mucoid impaction, and bronchial wall thickening as the earliest findings. Subsequent changes include areas of infiltrate, atelectasis, and hilar adenopathy. With advanced disease, segmental or lobar atelectasis, cyst formation, bronchiectasis , and pulmonary artery and right ventricular hypertrophy occur. Branching, fingerlike opacifications that represent mucoid impaction of dilated bronchi are characteristic.
Sinus CT studies are indicated in patients with significant sinus symptoms or nasal polyps in whom endoscopic sinus surgery is being considered. These studies almost always show persistent opacification of the paranasal sinuses.

This chest x-ray shows right lower lobe collapse. Findings are typical for CF but not specific.
By permission of the publisher. From Berman L: Atlas of Anesthesia: Critical Care . Edited by R Miller (series editor) and RR Kirby. Philadelphia, Current Medicine, 1997.

This chest x-ray of a man with cystic fibrosis shows increased lung markings consistent with bronchiectasis.
PHOTOSTOCK-ISRAEL/SCIENCE PHOTO LIBRARY

This CT scan shows greatly dilated bronchi throughout the lungs. Findings are typical for CF but not specific.
Pulmonary function tests are the best indicators of clinical status and response to therapy. In patients over 5 years of age, spirometry should be done routinely and at times of clinical decline. In infants, respiratory status can be monitored by using a raised-volume rapid thoracoabdominal compression technique, which generates a partial flow-volume curve. In children 3 to 6 years of age, the multiple breath washout procedure can be used to generate a lung clearance index as a measure of ventilation inhomogeneity ( 1 ).
Pulmonary function tests done by spirometry indicate
A reduction in forced vital capacity (FVC), forced expiratory volume in 1 second (FEV1), forced expiratory flow between 25% and 75% expired volume (FEF25-75), and FEV1/FVC ratio
An increase in residual volume and the ratio of residual volume to total lung capacity
Fifty percent of patients have evidence of reversible airway obstruction as shown by improvement in pulmonary function after administration of an inhaled bronchodilator.
Screening oropharyngeal or sputum cultures should be done at least 4 times/year, especially in patients not yet colonized with P. aeruginosa . Bronchoscopy/bronchoalveolar lavage is indicated when it is important to precisely define the patient’s lower airway microbial flora (eg, to direct antibiotic selection) or to remove inspissated mucus plugs.
Carrier screening
CF carrier screening is available in the United States and is recommended for couples who are planning a pregnancy or seeking prenatal care. If both potential parents carry a CFTR variant, prenatal screening of the fetus can be done by chorionic villus sampling or amniocentesis. Prenatal counseling in such cases is complicated by the wide phenotypic variability of CF and incomplete information on the clinical consequences of many of the CFTR variants that are identified through screening.
Diagnosis reference
1. Stanojevic S, Davis SD, Retsch-Bogart G, et al : Progression of lung disease in preschool patients with cystic fibrosis. Am J Respir Crit Care Med 195:1216–1225, 2017. doi: 10.1164/rccm.201610-2158OC
Treatment of Cystic Fibrosis
Comprehensive, multidisciplinary support
Antibiotics, inhaled medications to thin airway secretions, and physical maneuvers to clear airway secretions
Inhaled bronchodilators and sometimes corticosteroids for responders
Usually pancreatic enzyme and vitamin supplementation
High-calorie diet (sometimes requiring supplemental enteral tube feedings)
In patients with specific variants, CFTR modulators consisting of a CFTR potentiator or combination of CFTR correctors and a CFTR potentiator
Comprehensive and intensive therapy should be directed by an experienced physician working with a multidisciplinary team that includes other physicians, nurses, dietitians, physical and respiratory therapists, mental health professionals, pharmacists, and social workers. The goals of therapy are maintenance of normal nutritional status, prevention or aggressive treatment of pulmonary and other complications, encouragement of physical activity, and provision of psychosocial support. The treatment regimen is complex and may take up to 2 hours each day. With appropriate support, most patients can make an age-appropriate adjustment at home and school.
(See also the Cystic Fibrosis Foundation's comprehensive treatment guidelines for all age groups .)
Treatment of respiratory manifestations
Treatment of pulmonary manifestations centers on prevention of airway obstruction and prophylaxis against and control of pulmonary infections. Prophylaxis against pulmonary infections includes maintenance of pertussis , Haemophilus influenzae , varicella , Streptococcus pneumoniae , and measles immunity; annual influenza vaccination ; and COVID-19 vaccination in accordance with current recommendations prevention of respiratory syncytial virus infection has been shown to be safe, but efficacy has not been documented.
1, 2 ) and has been shown to slow the rate of decline in pulmonary function and to decrease the frequency of respiratory tract exacerbations ( 3 ).
Airway clearance measures consisting of postural drainage, percussion, vibration, and assisted coughing ( chest physiotherapy ) are recommended at the time of diagnosis and should be done on a regular basis. In older patients, alternative airway clearance measures, such as active cycle of breathing, autogenic drainage, positive expiratory pressure devices, and vest therapy (high-frequency chest wall oscillation), may be effective. Regular aerobic exercise is recommended; it may also help airway clearance. For patients with obstructive sleep apnea, continuous positive airway pressure may be beneficial.
For patients with reversible airway obstruction, bronchodilators may be given by inhalation. Corticosteroids by inhalation usually are not effective. Oxygen therapy is indicated for patients with severe pulmonary insufficiency and hypoxemia.
Mechanical ventilation or extracorporeal membrane oxygenation (ECMO) is typically not indicated for chronic respiratory failure . Their use is typically restricted to patients with good baseline status in whom acute reversible respiratory complications develop, in association with pulmonary surgery, or to patients in whom lung transplantation is imminent. Noninvasive positive pressure ventilation nasally or by face mask also can be beneficial.
Oral expectorants are sometimes used, but few data support their efficacy. Cough suppressants should be discouraged.
Pneumothorax can be treated with closed chest tube thoracostomy drainage. Open thoracotomy or thoracoscopy with resection of pleural blebs and mechanical abrasion of the pleural surfaces is effective in treating recurrent pneumothoraces .
Mild to moderate hemoptysis is treated with antibiotics (oral/aerosol or IV depending on severity of hemoptysis and severity of infection) and airway clearance. Massive or recurrent hemoptysis is treated by bronchial artery embolization or rarely by focal lung resection.
Oral corticosteroids are indicated in infants with prolonged bronchiolitis and in patients with refractory bronchospasm, allergic bronchopulmonary aspergillosis (ABPA), and inflammatory complications (eg, arthritis, vasculitis ). Long-term use of alternate-day corticosteroid therapy can slow the decline in pulmonary function, but because of corticosteroid-related complications, it is not recommended for routine use. Patients receiving corticosteroids must be closely monitored for evidence of diabetes and linear growth retardation.
Allergic bronchopulmonary aspergillosis is also treated with systemic corticosteroids and an oral antifungal medication.
CFTR modulators
CFTR corrector and potentiator medications are indicated for about 90% of the variants carried by patients with CF. CFTR modulators are not available for patients with class I frameshift and nonsense mutations.
CFTR variants. It may be used in patients 1 month of age and older who carry at least 1 copy of a specific variant potentiated by ivacaftor .
Lumacaftor, tezacaftor, and elexacaftor are small-molecule oral medications that partially correct the defective CFTR protein by altering protein misfolding in patients who carry the F508del variant or other specified variants.
The combination of lumacaftor and ivacaftor can be given to patients 1 year of age and older who carry 2 copies of the F508del variant.
The combination of tezacaftor and ivacaftor can be given to patients 6 years of age and older who carry 2 copies of the F508del variant or other specified variants.
The triple combination of elexacaftor, tezacaftor, and ivacaftor can be given to patients 2 years of age and older who carry at least 1 copy of the F508del variant or 1 copy of certain rare variants ( 4, 5 ).
These medications can improve pulmonary function, increase weight, improve exocrine pancreatic function, decrease the frequency of pulmonary exacerbations and hospitalizations, improve quality of life, and reduce and sometimes normalize sweat chloride concentrations ( 6 CFTR ivacaftor are considered to be highly effective modulator therapy.
Treatment and prevention of infections
For mild pulmonary exacerbations, S. aureus (MRSA), a course of oral trimethoprim P. aeruginosa
For moderate-to-severe pulmonary exacerbations, especially in patients colonized with P. aeruginosa
Eradication of chronic P. aeruginosa colonization is difficult. It has been shown, however, that early antibiotic treatment around the time the airways are initially infected with P. aeruginosa may be effective in eradicating the organism for some period of time. In patients who are chronically colonized with P. aeruginosa , antibiotics delivered by inhalation improve clinical parameters and possibly reduce the bacterial burden in the airways ( 7
Patients who have a clinically significant nontuberculous mycobacterium infection may require long-term therapy with a combination of oral, inhaled, and IV antibiotics.
Patients with allergic bronchopulmonary aspergillosis (ABPA) or lower airways aspergillus infection may require prolonged oral or IV therapy with an antifungal azole and/or systemic corticosteroids.
Treatment of gastrointestinal manifestations
Pancreatic enzyme replacement > 2,500 IU lipase/kg/meal or > 10,000 IU lipase/kg/day should be avoided because high enzyme dosages have been associated with fibrosing colonopathy. In patients with high enzyme requirements, acid suppression with an H2 blocker or proton pump inhibitor may improve enzyme effectiveness.
Diet therapy includes sufficient calories and protein to promote normal growth—30 to 50% more than the usual recommended dietary allowances may be required (see table Recommended Dietary Reference Intakes for Some Macronutrients ). Diet therapy also includes a normal-to-high total fat intake to increase the caloric density of the diet, a water-miscible multivitamin supplement in double the recommended daily allowance, supplementation with vitamin D3 (cholecalciferol) in patients with vitamin D deficiency or insufficiency, and salt supplementation during infancy and periods of thermal stress and increased sweating. Infants receiving broad-spectrum antibiotics and patients with liver disease and hemoptysis should be given additional supplemental vitamin K. Formulas containing protein hydrolysates and medium-chain triglycerides may be used instead of modified whole-milk formulas for infants with severe malabsorption. Glucose polymers and medium-chain triglyceride supplements can be used to increase caloric intake.
In patients who fail to maintain adequate nutritional status, enteral supplementation via gastrostomy or jejunostomy may improve growth and stabilize pulmonary function (see Overview of Nutritional Support ). The use of appetite stimulants to enhance growth may be helpful in some patients.
Treatment of other manifestations
Cystic fibrosis–related diabetes (CFRD) is caused by insulin insulin regimen, nutrition counseling, a diabetes self-management education program, and monitoring for microvascular complications. The plan should be carried out in conjunction with an endocrinologist and a dietitian with experience in treating both CF and diabetes.
Patients with symptomatic right heart failure should be treated with diuretics, salt restriction, and oxygen.
Recombinant human growth hormone (rhGH) may improve pulmonary function, increase height and weight and bone mineral content, and reduce the rate of hospitalization. However, because of the added cost and inconvenience, rhGH is not commonly used.
Surgery may be indicated for localized bronchiectasis or atelectasis that cannot be treated effectively with medications, nasal polyps, chronic rhinosinusitis, bleeding from esophageal varices secondary to portal hypertension, gallbladder disease, and intestinal obstruction due to a volvulus or an intussusception that cannot be medically reduced.
Liver transplantation has been done successfully in patients with end-stage liver disease.
Often, discussion of lung transplantation is needed. In considering transplantation, patients need to weigh the merits of longer survival with a transplant against the uncertainty of getting a transplant and the ongoing (but different) burden of living with an organ transplant. Bilateral cadaveric lung and live donor lobar transplantation has been done successfully in patients with advanced pulmonary disease. Combined liver-lung transplantation has been done for patients with end-stage liver and lung disease.
Bilateral lung transplantation for severe lung disease is becoming more routine and more successful with experience and improved techniques. Among adults with CF, median survival posttransplant is about 9 years.
Treatment references
1. Flume PA, O'Sullivan BP, Robinson KA, et al . Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007;176(10):957-969. doi:10.1164/rccm.200705-664OC
2. Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al . Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2013;187(7):680-689. doi:10.1164/rccm.201207-1160oe
3. Stahl M, Wielpütz MO, Ricklefs I, et al . Preventive Inhalation of Hypertonic Saline in Infants with Cystic Fibrosis (PRESIS). A Randomized, Double-Blind, Controlled Study. Am J Respir Crit Care Med 2019;199(10):1238-1248. doi:10.1164/rccm.201807-1203OC
4. Heijerman HGM, McKone EF, Downey DG, et al . Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial [published correction appears in Lancet 2020 May 30;395(10238):1694]. Lancet 2019;394(10212):1940-1948. doi:10.1016/S0140-6736(19)32597-8
5. Middleton PG, Mall MA, Dřevínek P, et al . Elexacaftor - Tezacaftor - Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med 2019;381(19):1809-1819. doi:10.1056/NEJMoa1908639
6. Taylor-Cousar JL, Robinson PD, Shteinberg M, Downey DG . CFTR modulator therapy: transforming the landscape of clinical care in cystic fibrosis. Lancet 2023;402(10408):1171-1184. doi:10.1016/S0140-6736(23)01609-4
7. Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al . Cystic Fibrosis Foundation pulmonary guideline. pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection. Ann Am Thorac Soc 2014;11(10):1640-1650. doi:10.1513/AnnalsATS.201404-166OC
Prognosis for Cystic Fibrosis
The course is largely determined by the degree of pulmonary involvement. Deterioration of pulmonary function over time, generally characterized by progressive bronchiectasis, leads to debilitation and eventually increases the risk for death, usually due to a combination of respiratory failure and cor pulmonale .
Prognosis has improved steadily over the past 5 decades, mainly because of early diagnosis and aggressive treatment before the onset of irreversible pulmonary changes. Median age at death in 2021 was 33.9 years. However, median predicted survival in the United States for children born in 2021 is age 65.6 years. Long-term survival is significantly better in patients without pancreatic insufficiency ( 1 ). Outcomes are also affected by CFTR variant profile, modifier genes, airway microbiology, sex, ambient temperature, exposure to air pollutants (including tobacco smoke), adherence to prescribed therapies, and socioeconomic status. The FEV1 , adjusted for age and sex, is the best predictor of survival. If health outcomes with CFTR modulator therapy are sustained, life expectancy can potentially increase even further.
End-of-life care
Patients and their families deserve sensitive discussions of prognosis and preferences for care throughout the course of illness, especially if pulmonary function progressively declines.
One mark of respect for patients living with CF is to ensure that they are given the information and opportunity to make life choices, including having a substantial hand in determining how and when to accept dying.
When appropriate, palliative care , including sufficient symptom management, should be offered to ensure peaceful end-of-life care. A useful strategy for the patient to consider is to accept a time-limited trial of fully aggressive treatment when needed, but to agree in advance to parameters that indicate when to stop aggressive measures (see Do-Not-Resuscitate (DNR) Orders and Portable Medical Orders ).
Prognosis reference
Cystic fibrosis is caused by carrying 2 variants of the gene for a protein called the cystic fibrosis transmembrane conductance regulator (CFTR), which regulates chloride, sodium, and bicarbonate transport across epithelial membranes.
The main complications involve the lungs, with damage to the small and large airways, chronic inflammation, and chronic and recurrent bacterial infections, particularly by Pseudomonas aeruginosa .
Other major consequences include pancreatic insufficiency, leading to malabsorption of nutrients and vitamins with consequent impaired growth and development, and, in older patients, a risk for developing diabetes.
Airway clearance measures (eg, postural drainage, percussion, vibration, assisted coughing), mucolytics, and airway hydrators are often started in early childhood; regular aerobic exercise is recommended.
Medications that correct or potentiate CFTR (CFTR modulators) can improve health outcomes for patients who have certain CFTR variants.
Antibiotics are given early in any pulmonary exacerbation; medication selection may be based on culture and sensitivity testing.
Diet should be supplemented with pancreatic enzymes, high-dose vitamins, and 30 to 50% more calories derived primarily from fat.
More Information
The following English-language resource may be useful. Please note that THE MANUAL is not responsible for the content of this resource.
Cystic Fibrosis Foundation: Age-specific care guidelines for cystic fibrosis

- Cystic Fibrosis
- Author: Girish D Sharma, MD, FCCP, FAAP; Chief Editor: Kenan Haver, MD more...
- Sections Cystic Fibrosis
- Practice Essentials
- Pathophysiology
- Epidemiology
- Patient Education
- Physical Examination
- Approach Considerations
- Prenatal, Neonatal, and Postnatal Testing
- Sweat Chloride Test
- Imaging Tests
- Nasal Potential Difference Measurement
- Pulmonary Function Testing
- Bronchoalveolar Lavage and Sputum Microbiology
- Immunoreactive Trypsinogen
- Contrast Barium Enema
- Diet and Exercise
- Surgical Management of Complications
- Special Populations
- Consultations and Long-Term Monitoring
- Medication Summary
- Enzymes, Pancreatic
- Bronchodilators
- Mucolytic Agents
- CFTR Potentiators and Correctors
- Antibiotics
- Questions & Answers
Cystic fibrosis (CF) is a disease of exocrine gland function that involves multiple organ systems but chiefly results in chronic respiratory infections, pancreatic enzyme insufficiency, and associated complications in untreated patients. Pulmonary involvement (see the image below) occurs in 90% of patients surviving the neonatal period. End-stage lung disease is the principal cause of death.

Signs and symptoms
Median age at diagnosis is 6-8 months; however, age at diagnosis varies widely. Clinical manifestations vary with the patient’s age at presentation.
Gastrointestinal (GI) symptoms may include the following:
Meconium ileus
Abdominal distention
Intestinal obstruction
Increased frequency of stools
Failure to thrive (despite adequate appetite)
Flatulence or foul-smelling flatus, steatorrhea
Recurrent abdominal pain
GI bleeding
Respiratory symptoms may include the following:
Recurrent wheezing
Recurrent pneumonia
Atypical asthma
Dyspnea on exertion
Genitourinary symptoms may include the following:
Undescended testicles or hydrocele
Delayed secondary sexual development
Physical signs depend on the degree of involvement of various organs and the progression of disease, as follows:
Nose – Rhinitis, nasal polyps
Pulmonary system – Tachypnea, respiratory distress with retractions, wheeze or crackles, cough (dry or productive of mucoid or purulent sputum), increased anteroposterior chest diameter, clubbing, cyanosis, hyperresonant chest on percussion
GI tract – Abdominal distention, hepatosplenomegaly, rectal prolapse, dry skin, cheilosis
See Clinical Presentation for more detail.
Requirements for a CF diagnosis include either positive genetic testing or positive sweat chloride test findings and 1 of the following:
Typical chronic obstructive pulmonary disease (COPD)
Documented exocrine pancreatic insufficiency
Positive family history (usually an affected sibling)
Parameters for the sweat chloride test are as follows:
The reference value is less than 40 mmol/L
A value higher than 60 mmol/L of chloride is consistent with CF
A value of 40-60 mmol/L is considered borderline, and the test must be repeated
In babies aged 3 months or younger, a value of 30-60 mEq/L is considered borderline and requires retesting [ 1 ]
Imaging studies that may be helpful include the following:
Radiography (chest, sinus, abdomen)
CT of the chest (not yet advised as a routine modality in CF)
Ultrasonography
Contrast barium enema
Additional tests that may be warranted are as follows:
Nasal potential difference measurement
Pulmonary function testing
Bronchoalveolar lavage
Sputum microbiology
Immunoreactive trypsinogen
See Workup for more detail.
The primary goals of CF treatment include the following:
Maintaining lung function as near to normal as possible by controlling respiratory infection and clearing airways of mucus
Administering nutritional therapy (ie, enzyme supplements, multivitamin and mineral supplements) to maintain adequate growth
Managing complications
Mild acute pulmonary exacerbations of CF can be treated successfully at home with the following measures:
Increasing the frequency of airway clearance
Inhaled bronchodilator treatment
Chest physical therapy and postural drainage
Increasing the dose of the mucolytic agent dornase alfa
Use of oral antibiotics (eg, fluoroquinolones)
Medications used to treat CF may include the following:
Pancreatic enzyme supplements
Multivitamins (including fat-soluble vitamins)
Nebulized, inhaled, oral, or intravenous antibiotics
Anti-inflammatory agents
Agents to treat associated conditions or complications (eg, insulin, bisphosphonates)
Agents devised to reverse abnormalities in chloride transport (eg, ivacaftor [ 2 ] )
Inhaled hypertonic saline
Surgical therapy may be required for the treatment of the following respiratory complications:
Respiratory – Pneumothorax, massive recurrent or persistent hemoptysis, nasal polyps, persistent and chronic sinusitis
GI – Meconium ileus, intussusception, gastrostomy tube placement for supplemental feeding, rectal prolapse
Lung transplantation is indicated for the treatment of end-stage lung disease. [ 3 ]
See Treatment and Medication for more detail.
Cystic fibrosis (CF) is the most common lethal inherited disease in white persons. [ 4 ] Cystic fibrosis is an autosomal recessive disorder, and most carriers of the gene are asymptomatic.
Cystic fibrosis is a disease of exocrine gland function that involves multiple organ systems but chiefly results in chronic respiratory infections, pancreatic enzyme insufficiency, and associated complications in untreated patients (see Clinical). Pulmonary involvement occurs in 90% of patients surviving the neonatal period. End-stage lung disease is the principal cause of death.
The diagnosis of cystic fibrosis is based on typical pulmonary manifestations, GI tract manifestations, a family history, and positive sweat chloride test results (see Workup). Newborn screening for cystic fibrosis is universally offered in the United States. As a result of the complex and multisystemic involvement of cystic fibrosis and the need for care by specialists, treatment and follow-up care at specialty centers with multidisciplinary care teams (ie, cystic fibrosis centers) is recommended (see Treatment).
Cystic fibrosis is caused by defects in the cystic fibrosis gene, which codes for a protein transmembrane conductance regulator ( CFTR ) that functions as a chloride channel and is regulated by cyclic adenosine monophosphate (cAMP). Mutations in the CFTR gene result in abnormalities of cAMP-regulated chloride transport across epithelial cells on mucosal surfaces.
Six classes of defects resulting from CFTR mutations have been described and are as follows [ 5 ] :
Complete absence of CFTR protein synthesis
Defective protein maturation and early degradation (caused by the most common mutation, ΔF508)
Disordered regulation (diminished ATP binding and hydrolysis)
Defective chloride conductance or channel gating
Diminished transcription due to promoter or splicing abnormality
Accelerated channel turnover from the cell surface
CFTR mutations have poor penetrance. This means that the genotype does not predict the pattern or severity of disease.
Defective CFTR results in decreased secretion of chloride and increased reabsorption of sodium and water across epithelial cells. The resultant reduced height of epithelial lining fluid and decreased hydration of mucus results in mucus that is stickier to bacteria, which promotes infection and inflammation. Secretions in the respiratory tract, pancreas, GI tract, sweat glands, and other exocrine tissues have increased viscosity, which makes them difficult to clear.
Most patients with cystic fibrosis have severe chronic lung disease and exocrine pancreatic insufficiency. Additional manifestations include the following:
Nasal polyposis
Pansinusitis
Rectal prolapse
Chronic diarrhea
Pancreatitis
Cholelithiasis
Cirrhosis or other forms of hepatic dysfunction
Sinus disease
The exact mechanism by which malfunctioning CFTR causes sinus disease is not completely understood. Chloride ions cannot be excreted, sodium is excessively absorbed, and water passively follows. This desiccates the mucosal surface and alters the viscosity of the normal mucus blanket, which alone can lead to obstruction of sinus ostia. [ 6 ]
Additional abnormalities exist in these patients, including ciliary dysfunction, increased inflammatory mediators, and increased colonization with Pseudomonas aeruginosa , all of which further impair normal sinus clearance and aeration. [ 7 ] Chronic sinus infection and inflammation are the net result.
Lung disease
Most deaths associated with cystic fibrosis result from progressive and end-stage lung disease. In individuals with cystic fibrosis, the lungs are normal in utero, at birth, and after birth, before the onset of infection and inflammation (except possibly for the presence of dilated submucosal gland ducts in the airways). Shortly after birth, many persons with cystic fibrosis acquire a lung infection, which incites an inflammatory response. Infection becomes established with a distinctive bacterial flora. A repeating cycle of infection and neutrophilic inflammation develops.
Cleavage of complement receptors CR1 and C3bi and immunoglobulin G (IgG) by neutrophil elastase (NE) results in failure of opsonophagocytosis, leading to bacterial persistence. NE also causes production of the neutrophil chemoattractant interleukin (IL)–8 from epithelial cells and elastin degradation and acts as secretogogue, thereby contributing to persistence of inflammation and infection, structural damage, impaired gas exchange, and, ultimately, end-stage lung disease and early death.
One study reported that exposure to secondhand smoke adversely affects both cross-sectional and longitudinal measures of lung function in individuals with cystic fibrosis. [ 8 ] Variations in CFTR and a cystic fibrosis–modifier gene ( TGFβ1 ) amplify the negative effects of secondhand smoke exposure.
Intestinal disease
Defects in CFTR lead to reduced chloride secretion with water following into the gut. This may result in meconium ileus at birth and in distal intestinal obstruction syndrome (DIOS) later in life.
In addition, other pathologic disorders complicate the simple relationship between the apical chloride and water secretion and the disease. The pancreatic insufficiency decreases the absorption of intestinal contents.
Mechanical problems associated with inflammation, scarring, and strictures may predispose the patient to sludging of intestinal contents, leading to intestinal obstruction by fecal impaction or to intussusception. Adhesions may form, leading to complete obstruction. A complete obstruction may require resection, leading to loss of absorptive epithelium of the distal ileum.
The meconium of fetuses with cystic fibrosis and meconium ileus has increased viscosity and decreased water content compared with those of healthy controls. The developmental sequence of mucin secretion in the fetal intestine is not fully understood, although the CFTR ion channel defect possibly leads to dehydration of intraluminal contents.
Meconium in patients with meconium ileus also has higher protein and lower carbohydrate concentration than that in control populations. Albumin is the major protein in the meconium of infants with meconium ileus, and is present in concentrations 5-10 times higher than normal. [ 9 ] In addition, there is a significant increase in the liver's production of intraluminal glutamyltranspeptidase (GGTP) and 5'-nucleotidase, which enters the meconium and promotes meconium ileus.
The addition of albumin to normal meconium makes it viscid; the addition of pancreatic protease liquefies the viscid mass. This led to the belief that pancreatic insufficiency played a central role in the pathogenesis of meconium ileus, although pancreatic insufficiency is not the sole cause of abnormal meconium in meconium ileus. In 1988, however, Lands et al reported 2 infants with cystic fibrosis and meconium ileus, aged 9 and 11 months, who displayed no clinical evidence of pancreatic insufficiency. [ 10 ]
In the murine model of cystic fibrosis, developed in 1992, newborn mice had severe intestinal obstruction at birth with minimal pulmonary or pancreatic involvement. These animal studies support the concept that meconium ileus may occur in patients with sufficient pancreatic activity. The lack of concordance between meconium ileus and severity of pancreatic disease suggests that intraluminal intestinal factors contribute to meconium ileus development.
Abnormal intestinal motility may also contribute to meconium ileus development. Some patients with cystic fibrosis have prolonged small intestinal transit times. Diseases other than cystic fibrosis in which there is abnormal gut motility (eg, Hirschsprung disease , chronic intestinal pseudo-obstruction) have been associated with meconium ileus–like disease, suggesting that decreased peristalsis may allow increased resorption of water, thus favoring meconium ileus development.
Pancreatic disease
As a part of normal digestion, stomach acid is neutralized by pancreatic bicarbonate, leading to the optimal pH for pancreatic enzyme action. Reduced bicarbonate secretion in response to secretin stimulation has been demonstrated in patients with cystic fibrosis with both pancreatic insufficiency and sufficiency. Reduced bicarbonate secretion affects the digestion so that neither endogenous nor exogenous pancreatic enzymes can work at their optimal pH.
Other factors, such as reduced water content of secretions, precipitation of proteins, and plugging of ductules and acini, prevent the pancreatic enzymes from reaching the gut. Autodigestion of the pancreas occasionally leads to pancreatitis.
Most patients with cystic fibrosis (90-95%) have pancreatic enzyme insufficiency and present with digestive symptoms and/or failure to thrive early in life. Onset of pancreatic insufficiency varies, however, and may occur in patients older than 6 months. Some patients never develop pancreatic insufficiency.
Patients with pancreatic insufficiency typically present with poor weight gain in association with frequent stools that are malodorous, greasy, and associated with flatulence and colicky pain after feeding. The combination of increased energy intake demand at baseline, the added energy intake demand of chronic disease, difficulty sustaining energy uptake because of malabsorption, and anorexia associated with ongoing lung inflammation leads to poor weight gain.
Pancreatic insufficiency predisposes patients to poor absorption of fat-soluble vitamins A, D, E, and K. Symptomatic deficiency of any of these vitamins can occur before diagnosis or as a later complication of the disease.
Liver disease
Absence of functional CFTR in epithelial cells lining the biliary ductules leads to reduced secretion of chloride and reduction in passive transport of water and chloride, resulting in increased viscosity of bile. The biliary ductules may be plugged with secretions. If this process is extensive, obstructive cirrhosis complicated by esophageal varices, splenomegaly, and hypersplenism may occur.
Secondary involvement of the liver may also occur because of involvement of other organs. For example, malnutrition may be associated with hepatic steatosis, and right heart failure caused by chronic hypoxia may result in passive congestion of the liver.
Gallstones are more prevalent in patients with cystic fibrosis than in age-matched control subjects. As many as 15% of young adults with cystic fibrosis have gallstones, irrespective of the status of their pancreatic function. Abnormal mucin in the gallbladder and malabsorption of bile acids in a patient with PI result in a higher frequency of gallstones.
Urogenital disease
Congenital absence of vas deferens may result in male infertility. Undescended testicles or hydroceles may be present in boys. Fertility is possibly decreased in females. Amenorrhea may occur in females with severe nutritional or pulmonary involvement.
Cystic fibrosis is an autosomal recessive disease caused by defects in the CFTR gene, which encodes for a protein that functions as a chloride channel, and also regulates the flow of other ions across the apical surface of epithelial cells. In 1989, the CF locus was localized through linkage analysis to the long arm of human chromosome 7, band q31. [ 11 ]
Thus far, 1893 CFTR mutations have been identified. [ 12 ] Half of affected individuals of northern European descent are homozygous for the ΔF508 mutation, which is the deletion of a single phenylalanine residue at amino acid 508 of the CFTR gene (a class II defect). Another 25%-30% have one copy of ΔF508 plus another mutation. [ 13 ]
Certain alleles cluster with increased frequency in specific populations. For example, W1282X is common in Ashkenazi Jews, and A455E is common both in Dutch people and in individuals from northern Quebec. Δ1152H is the third most prevalent allele in Ashkenazi and other ethnic Jewish groups. The prevalence of Δ1152 mutation in Jewish populations comprises 5.2% of all CFTR mutations.
CFTR mutations result in abnormalities of cAMP-regulated chloride transport across epithelial cells on mucosal surfaces. The failure of chloride conductance by epithelial cells and associated water transport abnormalities result in viscid secretions in the respiratory tract, pancreas, GI tract, sweat glands, and other exocrine tissues. Increased viscosity of these secretions makes them difficult to clear.
Genotype-phenotype correlation demonstrates that ΔF508 homozygosity nearly always confers a pancreatic exocrine insufficiency. Individuals with 1 or 2 copies of missense mutations (eg, R117H) tend to be pancreatic sufficient and have milder disease.
The incidence of meconium ileus is higher in patients who are homozygous for ΔF508 or who have ΔF508 plus G542X . Conversely, not all patients with these genotypes have meconium ileus, so other non -CFTR factors must be involved in meconium ileus pathogenesis.
The incomplete correlation of genotype with phenotype suggests either an environmental component of organ dysfunction or modifying genes that are only recently being characterized. [ 14 ] The role of modifier genes is supported by the fact that neonates with cystic fibrosis who have intestinal obstruction most commonly have abnormalities in 2 or more CFTR modifier genes. In contrast, older children develop obstruction mostly as a result of environmental factors, such as introduction of pancreatic enzymes causing a stricture. [ 15 , 16 ]
Studies in murine CF models have shown an increase in mast cells and neutrophils as part of the immune response. For example, the KITL gene plays a vital role in the differentiation of mast cells, as demonstrated by a decreased expression of MCPT2 . Another focus includes the proteins selectin and intercellular adhesion molecule–1 (ICAM-1), which facilitate neutrophil extravasation. Neutrophils and mast cells release proteases, prostaglandins, and histamine, influencing mucus production.
A research model found in CFTR- knockout gene mice highlighted the importance of MCLCA3 expression in goblet cells. This gene influences mucus production, among other activities, and its expression was noted to be diminished in these mice. Correction of this deficiency increased survival and decreased intestinal disease. In humans, this finding may translate to applications such as correcting modifier genes (eg, HCLCA1 ) in order to improve outcomes in patients with CF. [ 17 ]
Additional genetic modifiers include a 129/Sv allelic contribution in mice that yields a milder inflammatory response in CF and is potentially linked to chromosomes 1, 9, and 10. The regulation of these genes and processes helps explain the range of phenotypic variability in similar genetic mutations.
Cystic fibrosis is an autosomal-recessive disease. Its estimated heterozygote frequency in white people is up to 1 in 20. Each offspring of 2 heterozygote parents has a 25% chance of developing cystic fibrosis.
Cystic fibrosis is the most common lethal hereditary disease in the white population. In the United States, the prevalence is as follows:
Whites of northern European origin - 1 case per 3,200-3,500 population
Hispanics - 1 case per 9,200-9,500 population
African Americans - 1 case per 15,000-17,000 population
Asian Americans - 1 case per 31,000 population
The worldwide incidence varies from 1 per 377 live births in parts of England to 1 per 90,000 Asian live births in Hawaii. The higher frequency in Asian American or African American populations compared with native Asians or Africans reflects white admixture. [ 18 ]
Race demographics
The distribution of CFTR mutations varies according to the background of patients; for example, ΔF508 is the most common mutation found in the white population of northern European origin. Variability in clinical features between people of different races with same genotype has not been reported.
Clinical manifestations are similar in black and white populations, except that a poorer nutritional status is observed in black patients. Black patients with cystic fibrosis are younger at diagnosis and have poorer nutritional status and pulmonary function than white patients with cystic fibrosis. Whether this is genetic or due to socioeconomic factors is unclear; low socioeconomic status is associated with significantly worse pulmonary outcomes in patients with cystic fibrosis.
Sex demographics
Compared with males, females with cystic fibrosis have greater deterioration of pulmonary function with increasing age and younger mean age at death. [ 19 ] Although it has been suggested that the increase in hormone secretion with puberty in females may interfere with the defense mechanisms of the immune system, thereby promoting progressive pulmonary involvement, the immune system in patients with cystic fibrosis is fundamentally intact.
Worldwide, the median survival age in patients with cystic fibrosis varies from country to country; it is highest in the United States. [ 20 ] Median survival age is 36.9 years, but progress in medical and surgical treatment options have improved the prognosis over the last few decades. An individual with cystic fibrosis born in the United States today is expected to survive longer than 40 years. [ 21 ] The median survival age is higher in males than in females.
With current treatment strategies, 80% of patients should reach adulthood. Nevertheless, cystic fibrosis remains a life-limiting disease, and a cure for the disease remains elusive.
The clinical presentation, age at diagnosis, severity of symptoms, and rate of disease progression in the organs involved widely vary. Sweat abnormalities may result in heat stroke and salt depletion, especially in infants. Mucocele and mucopyocele associated with chronic sinusitis and nasal polyps can cause erosion of the sinus wall, resulting in CNS complications from the space-occupying effect of mucopyocele or from associated complications. [ 22 ]
GI tract complications include pancreatic involvement. Pancreatic tissue damage leads to diabetes mellitus in 8-12% of patients older than 25 years. Excessive administration of exogenous pancreatic enzymes can result in fibrosing colonopathy. Intestinal complications range from meconium ileus with associated complications during the neonatal period (12% of neonates with cystic fibrosis) to distal intestinal obstruction syndrome, rectal prolapse, peptic ulcer, and gastroesophageal reflux .
Liver involvement may result in a fatty liver (30-60% of patients), focal biliary cirrhosis, multinodular biliary cirrhosis, and associated portal hypertension. Portal hypertension occasionally causes death through esophageal varices. The prevalence of cholecystitis and gallstones is higher in patients with cystic fibrosis than in other individuals.
Delayed puberty and reduced fertility are other complications; most males are azoospermic because of agenesis of the vas deferens. Female fertility is probably only mildly impaired, and many successful pregnancies have been reported in women with cystic fibrosis.
Severity of pulmonary disease determines prognosis and ultimate outcome. Pulmonary involvement is progressive: beginning as bronchitis, bronchiolitis, and then bronchiectasis, pulmonary involvement leads to cor pulmonale and end-stage lung disease. Cause of death is generally respiratory failure and cor pulmonale.
A review of 6750 deaths due to cystic fibrosis in England and Wales from 1959-2008 reported that female sex and low socioeconomic status are associated with poorer outcomes than male sex and high socioeconomic status. [ 23 ]
A study of 1517 patients with cystic fibrosis who were registered with the UK Cystic Fibrosis Registry showed that lower muscle mass, shorter stature, and a low body mass index are associated with increased mortality. [ 24 ]
In a prospective observational study of 3142 patients from the Cystic Fibrosis Foundation Registry, weight for age percentile at 4 years of age was associated with improved clinical outcomes including lung function, fewer complications of cystic fibrosis and better survival through the age of 18. [ 25 ]
Provide counseling at the time of initial diagnosis, including information regarding inheritance and risk for recurrence in subsequent pregnancies, and instruct patients and parents regarding appropriate airway clearance technique and the need for chest physical therapy. Also, instruct patients and parents regarding the use of various drug delivery devices, such as valved holding chambers, and nebulizers, and the methods for modifying the pancreatic enzyme dosage.
Discuss when to contact cystic fibrosis center personnel (eg, for acute pulmonary exacerbation or complications) with patients and parents, and be prepared to counsel families regarding the impact of the diagnosis on the emotional life of parents, siblings, and members of the extended family.
LeGrys VA, Yankaskas JR, Quittell LM, Marshall BC, Mogayzel PJ Jr. Diagnostic sweat testing: the Cystic Fibrosis Foundation guidelines. J Pediatr . 2007 Jul. 151(1):85-9. [QxMD MEDLINE Link] .
Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med . 2011 Nov 3. 365(18):1663-72. [QxMD MEDLINE Link] .
Yankaskas JR, Mallory GB Jr. Lung transplantation in cystic fibrosis: consensus conference statement. Chest . 1998 Jan. 113(1):217-26. [QxMD MEDLINE Link] .
Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med . 1996 Nov. 154(5):1229-56. [QxMD MEDLINE Link] .
Rowe SM, Clancy JP. Advances in cystic fibrosis therapies. Curr Opin Pediatr . 2006 Dec. 18(6):604-13. [QxMD MEDLINE Link] .
Rutland J, Cole PJ. Nasal mucociliary clearance and ciliary beat frequency in cystic fibrosis compared with sinusitis and bronchiectasis. Thorax . 1981. 36:654-658.
Hauber HP, Manoukian JJ, Nguyen LHP. Increased expression of interleukin-9, interleukin-9 receptor, and the calcium-activated chloride channel hCLCA1 in the upper airways of patients with cystic fibrosis. Laryngoscope . 2003. 113:1037-1042.
Collaco JM, Vanscoy L, Bremer L, et al. Interactions between secondhand smoke and genes that affect cystic fibrosis lung disease. JAMA . 2008 Jan 30. 299(4):417-24. [QxMD MEDLINE Link] .
GREEN MN, CLARKE JT, SHWACHMAN H. Studies in cystic fibrosis of the pancreas; protein pattern in meconium ileus. Pediatrics . 1958 Apr. 21(4):635-41. [QxMD MEDLINE Link] .
Gross R. Intestinal Obstruction in the Newborn Arising from Meconium Ileus. The surgery of infants and childhood . 1953. 175-191.
Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science . 1989 Sep 8. 245(4922):1059-65. [QxMD MEDLINE Link] .
Cystic Fibrosis Genetic Analysis Consortium. Cystic fibrosis mutation database-. CFMDB Statistics. Available at http://www.genet.sickkids.on.ca/cftr/StatisticsPage.html . Accessed: October 14, 2011.
Julian Zielenski, Anluan O'Brien and Lap-Chee Tsui. Cystis Fibrosis Mutation Database. Cystic Fibrosis Genetic Analysis Consortium. Available at http://www.genet.sickkids.on.ca/cftr/StatisticsPage.html . Accessed: June 24, 2008.
Gambardella S, Biancolella M, D'Apice MR, et al. Gene expression profile study in CFTR mutated bronchial cell lines. Clin Exp Med . 2006 Dec. 6(4):157-65. [QxMD MEDLINE Link] .
Chaudry G, Navarro OM, Levine DS, Oudjhane K. Abdominal manifestations of cystic fibrosis in children. Pediatr Radiol . 2006 Mar. 36(3):233-40. [QxMD MEDLINE Link] .
Blackman SM, Deering-Brose R, McWilliams R, et al. Relative contribution of genetic and nongenetic modifiers to intestinal obstruction in cystic fibrosis. Gastroenterology . 2006 Oct. 131(4):1030-9. [QxMD MEDLINE Link] . [Full Text] .
Young FD, Newbigging S, Choi C, Keet M, Kent G, Rozmahel RF. Amelioration of cystic fibrosis intestinal mucous disease in mice by restoration of mCLCA3. Gastroenterology . 2007 Dec. 133(6):1928-37. [QxMD MEDLINE Link] .
National Institutes of Health Consensus. Genetic testing for cystic fibrosis. National Institutes of Health Consensus Development Conference Statement on genetic testing for cystic fibrosis. Arch Intern Med . 1999 Jul 26. 159(14):1529-39. [QxMD MEDLINE Link] .
Boat TF. Cystic fibrosis. Nelson Textbook of Pediatrics . Philadelphia, Pa: WB Saunders Co; 2000. 1315-1327.
Cystic Fibrosis Foundation. Fibrosis Foundation Patient registry Annual Report 2008 . Bethesda, MD: Cystic Fibrosis Foundation; 2009.
Elborn JS, Shale DJ, Britton JR. Cystic fibrosis: current survival and population estimates to the year 2000. Thorax . 1991 Dec. 46(12):881-5. [QxMD MEDLINE Link] . [Full Text] .
Sharma GD, Doershuk CF, Stern RC. Erosion of the wall of the frontal sinus caused by mucopyocele in cystic fibrosis. J Pediatr . 1994 May. 124(5 Pt 1):745-7. [QxMD MEDLINE Link] .
Barr HL, Britton J, Smyth AR, Fogarty AW. Association between socioeconomic status, sex, and age at death from cystic fibrosis in England and Wales (1959 to 2008): cross sectional study. BMJ . 2011 Aug 23. 343:d4662. [QxMD MEDLINE Link] .
Fogarty AW, Britton J, Clayton A, Smyth A. Are measures of body habitus associated with mortality in cystic fibrosis?. Chest . 2012 Feb 23. [QxMD MEDLINE Link] .
Yen EH, Quinton H, Borowitz D. Better Nutritional Status in Early Childhood Is Associated with Improved Clinical Outcomes and Survival in Patients with Cystic Fibrosis. J Pediatr . 2012 Oct 11. [QxMD MEDLINE Link] .
Berk DR, Ciliberto HM, Sweet SC, Ferkol TW, Bayliss SJ. Aquagenic wrinkling of the palms in cystic fibrosis: comparison with controls and genotype-phenotype correlations. Arch Dermatol . 2009 Nov. 145(11):1296-9. [QxMD MEDLINE Link] .
Kelly A, Schall J, Stallings VA, Zemel BS. Trabecular and cortical bone deficits are present in children and adolescents with cystic fibrosis. Bone . 2016 Apr 29. [QxMD MEDLINE Link] .
Boggs W. Bone Deficits Common in Children With Cystic Fibrosis. Reuters Health Information. Available at http://www.medscape.com/viewarticle/863370 . May 18, 2016; Accessed: June 8, 2016.
Zielenski J, Patrizio P, Corey M, et al. CFTR gene variant for patients with congenital absence of vas deferens. Am J Hum Genet . 1995 Oct. 57(4):958-60. [QxMD MEDLINE Link] . [Full Text] .
Vande Velde S, Van Biervliet S, Robberecht E. Cystic fibrosis presenting as diabetes insipidus unresponsive to desmopressin. Acta Gastroenterol Belg . 2007 Jul-Sep. 70(3):300-1. [QxMD MEDLINE Link] .
[Guideline] Comeau AM, Accurso FJ, White TB, et al. Guidelines for implementation of cystic fibrosis newborn screening programs: Cystic Fibrosis Foundation workshop report. Pediatrics . 2007 Feb. 119(2):e495-518. [QxMD MEDLINE Link] . [Full Text] .
Hale JE, Parad RB, Comeau AM. Newborn screening showing decreasing incidence of cystic fibrosis. N Engl J Med . 2008 Feb 28. 358(9):973-4. [QxMD MEDLINE Link] .
Cystic Fibrosis Foundation., Borowitz D, Parad RB, Sharp JK, Sabadosa KA, Robinson KA, et al. Cystic Fibrosis Foundation practice guidelines for the management of infants with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome during the first two years of life and beyond. J Pediatr . 2009 Dec. 155 (6 Suppl):S106-16. [QxMD MEDLINE Link] .
Farrell PM, Koscik RE. Sweat chloride concentrations in infants homozygous or heterozygous for F508 cystic fibrosis. Pediatrics . 1996 Apr. 97(4):524-8. [QxMD MEDLINE Link] .
Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, Derichs N, et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr . 2017 Feb. 181S:S4-S15.e1. [QxMD MEDLINE Link] .
Leonidas JC, Berdon WE, Baker DH, Santulli TV. Meconium ileus and its complications. A reappraisal of plain film roentgen diagnostic criteria. Am J Roentgenol Radium Ther Nucl Med . 1970 Mar. 108(3):598-609. [QxMD MEDLINE Link] .
Sanders DB, Li Z, Brody AS, Farrell PM. Chest CT Scores of Severity are Associated with Future Lung Disease Progression in Children with CF. Am J Respir Crit Care Med . 2011 Jul 7. [QxMD MEDLINE Link] .
Rosenow T, Ramsey K, Turkovic L, Murray CP, Mok LC, Hall GL, et al. Air trapping in early cystic fibrosis lung disease-Does CT tell the full story?. Pediatr Pulmonol . 2017 Jul 6. [QxMD MEDLINE Link] .
Dicke JM, Crane JP. Sonographically detected hyperechoic fetal bowel: significance and implications for pregnancy management. Obstet Gynecol . 1992 Nov. 80(5):778-82. [QxMD MEDLINE Link] .
Savant AP, McColley SA. Cystic fibrosis year in review 2016. Pediatr Pulmonol . 2017 Aug. 52 (8):1092-1102. [QxMD MEDLINE Link] .
Smith L, Marshall H, Aldag I, Horn F, Collier G, Hughes D, et al. Longitudinal Assessment of Children with Mild CF Using Hyperpolarised Gas Lung MRI and LCI. Am J Respir Crit Care Med . 2017 Jun 29. [QxMD MEDLINE Link] .
Marshall H, Horsley A, Taylor CJ, Smith L, Hughes D, Horn FC, et al. Detection of early subclinical lung disease in children with cystic fibrosis by lung ventilation imaging with hyperpolarised gas MRI. Thorax . 2017 Aug. 72 (8):760-762. [QxMD MEDLINE Link] .
US Food and Drug Administration. FDA allows marketing of four "next generation" gene sequencing devices [news release]. November 19, 2013. Available at http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm375742.htm . Accessed: November 25, 2013.
Brooks M. FDA OKs 'next-gen' gene sequencing devices for clinical use. Medscape Medical News . November 20, 2013. [Full Text] .
Rosenfeld M, Allen J, Arets BH, Aurora P, Beydon N, Calogero C, et al. An official American Thoracic Society workshop report: optimal lung function tests for monitoring cystic fibrosis, bronchopulmonary dysplasia, and recurrent wheezing in children less than 6 years of age. Ann Am Thorac Soc . 2013 Apr. 10(2):S1-S11. [QxMD MEDLINE Link] .
Filburn AG, Lumeng CN, Nasr SZ. Infant pulmonary function testing guides therapy in cystic fibrosis lung disease. Respiratory Medicine CME . 2011. 4:17-19.
Ren CL, Brucker JL, Rovitelli AK, Bordeaux KA. Changes in lung function measured by spirometry and the forced oscillation technique in cystic fibrosis patients undergoing treatment for respiratory tract exacerbation. Pediatr Pulmonol . 2006 Apr. 41(4):345-9. [QxMD MEDLINE Link] .
Taylor-Robinson D, Whitehead M, Diderichsen F, Olesen HV, Pressler T, Smyth RL, et al. Understanding the natural progression in %FEV1 decline in patients with cystic fibrosis: a longitudinal study. Thorax . 2012 May 3. [QxMD MEDLINE Link] .
Davies JC, Cunningham S, Alton EW, Innes JA. Lung clearance index in CF: a sensitive marker of lung disease severity. Thorax . 2008 Feb. 63(2):96-7. [QxMD MEDLINE Link] .
Moran A, Pekow P, Grover P, et al. Insulin therapy to improve BMI in cystic fibrosis-related diabetes without fasting hyperglycemia: results of the cystic fibrosis related diabetes therapy trial. Diabetes Care . 2009 Oct. 32(10):1783-8. [QxMD MEDLINE Link] . [Full Text] .
Lum S, Gustafsson P, Ljungberg H, Hülskamp G, Bush A, Carr SB. Early detection of cystic fibrosis lung disease: multiple-breath washout versus raised volume tests. Thorax . 2007 Apr. 62(4):341-7. [QxMD MEDLINE Link] .
Owens CM, Aurora P, Stanojevic S, Bush A, Wade A, Oliver C. Lung Clearance Index and HRCT are complementary markers of lung abnormalities in young children with CF. Thorax . 2011 Jun. 66(6):481-8. [QxMD MEDLINE Link] .
Steven LC, Gavel G, Young D, Carachi R. Immunoreactive trypsin levels in neonates with meconium ileus. Pediatr Surg Int . 2006 Mar. 22(3):236-9. [QxMD MEDLINE Link] .
Santulli TV, Blanc WA. Congenital atresia of the intestine: pathogenesis and treatment. Ann Surg . 1961 Dec. 154:939-48. [QxMD MEDLINE Link] . [Full Text] .
Shinohara T, Tsuda M, Koyama N. Management of meconium-related ileus in very low-birthweight infants. Pediatr Int . 2007 Oct. 49(5):641-4. [QxMD MEDLINE Link] .
Yang C, Montgomery M. Dornase alfa for cystic fibrosis. Cochrane Database Syst Rev . 2018 Sep 6. 9:CD001127. [QxMD MEDLINE Link] .
Sagel SD, Khan U, Jain R, Graff G, Daines CL, Dunitz JM, et al. Effects of an Antioxidant-enriched Multivitamin in Cystic Fibrosis. A Randomized, Controlled, Multicenter Clinical Trial. Am J Respir Crit Care Med . 2018 Sep 1. 198 (5):639-647. [QxMD MEDLINE Link] .
Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med . 2015 May 17. [QxMD MEDLINE Link] . [Full Text] .
Taylor-Cousar JL, Munck A, McKone EF, van der Ent CK, Moeller A, Simard C, et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med . 2017 Nov 23. 377 (21):2013-2023. [QxMD MEDLINE Link] .
Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, Tullis E, et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N Engl J Med . 2017 Nov 23. 377 (21):2024-2035. [QxMD MEDLINE Link] .
Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med . 2006 Jan 19. 354(3):241-50. [QxMD MEDLINE Link] .
Wark P, McDonald VM. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst Rev . 2018 Sep 27. 9:CD001506. [QxMD MEDLINE Link] .
Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med . 2006 Jan 19. 354(3):229-40. [QxMD MEDLINE Link] .
Flume PA, O'Sullivan BP, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med . 2007 Nov 15. 176(10):957-69. [QxMD MEDLINE Link] .
Yu H, Burton B, Huang CJ, Worley J, Cao D, Johnson JP Jr, et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros . 2012 May. 11(3):237-45. [QxMD MEDLINE Link] .
Rowe, Steven M., et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. New England Journal of Medicine . 2017 Nov 23. 377 (21):2024-2035. [QxMD MEDLINE Link] . [Full Text] .
Taylor-Cousar JL, et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med . 2017 Nov 23. 377 (21):2013-2023. [QxMD MEDLINE Link] . [Full Text] .
Symdeko (tezacaftor/ivacaftor) [package insert]. Boston, MA: Vertex Pharmaceuticals, Inc. June 2019. Available at [Full Text] .
Cheng K, Ashby D, Smyth RL. Oral steroids for long-term use in cystic fibrosis. Cochrane Database Syst Rev . 2011 Oct 5. CD000407. [QxMD MEDLINE Link] .
Aitken ML, Bellon G, De Boeck K, Flume PA, Fox HG, Geller DE, et al. Long-term inhaled dry powder mannitol in cystic fibrosis: an international randomized study. Am J Respir Crit Care Med . 2012 Mar 15. 185(6):645-52. [QxMD MEDLINE Link] .
FDA approves TOBI Podhaler to treat a type of bacterial lung infection in cystic fibrosis patients. FDA News Release. March 22, 2013. Available at http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm345123.htm . Accessed: April 1, 2013.
Lewis R. Cystic fibrosis: bisphosphonate ups bone density in children. Medscape Medical News . June 6, 2013. [Full Text] .
Bianchi ML, Assael B, Dubini A, et al. Treatment of low bone density in young people with cystic fibrosis: a multicentre, prospective, open-label observational study of calcium and calcifediol followed by a randomised placebo-controlled trial of alendronate. Lancet Resp Med . 2013 Jun 2. [Epub ahead of print].
Best C, Brearley A, Gaillard P, et al. A pre-post retrospective study of patients with cystic fibrosis and gastrostomy tubes. J Pediatr Gastroenterol Nutr . 2011 Oct. 53(4):453-8. [QxMD MEDLINE Link] .
Schwarzenberg SJ, Hempstead SE, McDonald CM, Powers SW, Wooldridge J, Blair S, et al. Enteral tube feeding for individuals with cystic fibrosis: Cystic Fibrosis Foundation evidence-informed guidelines. J Cyst Fibros . 2016 Nov. 15 (6):724-735. [QxMD MEDLINE Link] .
Liou TG, Adler FR, Cox DR, Cahill BC. Lung transplantation and survival in children with cystic fibrosis. N Engl J Med . 2007 Nov 22. 357(21):2143-52. [QxMD MEDLINE Link] . [Full Text] .
Allen J, Visner G. Lung transplantation in cystic fibrosis--primum non nocere?. N Engl J Med . 2007 Nov 22. 357(21):2186-8. [QxMD MEDLINE Link] .
Al-Saleh S, Dell SD, Grasemann H, Yau YC, Waters V, Martin S, et al. Sputum induction in routine clinical care of children with cystic fibrosis. J Pediatr . 2010 Dec. 157(6):1006-1011.e1. [QxMD MEDLINE Link] .
Robinson KA, Odelola OA, Saldanha IJ, McKoy NA. Palivizumab for prophylaxis against respiratory syncytial virus infection in children with cystic fibrosis. Cochrane Database Syst Rev . 2012 Feb 15. 2:CD007743. [QxMD MEDLINE Link] .
Kazmerski TM, Borrero S, Tuchman LK, Weiner DJ, Pilewski JM, Orenstein DM, et al. Provider and Patient Attitudes Regarding Sexual Health in Young Women With Cystic Fibrosis. Pediatrics . 2016 Jun. 137 (6): [QxMD MEDLINE Link] .
Konstan MW, McKone EF, Moss RB, Marigowda G, Tian S, Waltz D, et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respir Med . 2017 Feb. 5 (2):107-118. [QxMD MEDLINE Link] .
Morgan WJ, Wagener JS, Pasta DJ, Millar SJ, VanDevanter DR, Konstan MW, et al. Relationship of Antibiotic Treatment to Recovery after Acute FEV 1 Decline in Children with Cystic Fibrosis. Ann Am Thorac Soc . 2017 Jun. 14 (6):937-942. [QxMD MEDLINE Link] .
Smyth AR, Bhatt J. Once-daily versus multiple-daily dosing with intravenous aminoglycosides for cystic fibrosis. Cochrane Database Syst Rev . 2012 Feb 15. 2:CD002009. [QxMD MEDLINE Link] .
Schuster A, Haliburn C, Döring G, Goldman MH. Safety, efficacy and convenience of colistimethate sodium dry powder for inhalation (Colobreathe DPI) in patients with cystic fibrosis: a randomised study. Thorax . 2012 Dec 4. [QxMD MEDLINE Link] .
Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA . 2003 Oct 1. 290(13):1749-56. [QxMD MEDLINE Link] .
Nick JA, Moskowitz SM, Chmiel JF, et al. Azithromycin May Antagonize Inhaled Tobramycin When Targeting Pseudomonas aeruginosa in Cystic Fibrosis. Ann Am Thorac Soc . 2014 Mar. 11(3):342-50. [QxMD MEDLINE Link] .
Taccetti G, Bianchini E, Cariani L, Buzzetti R, Costantini D, Trevisan F, et al. Early antibiotic treatment for Pseudomonas aeruginosa eradication in patients with cystic fibrosis: a randomised multicentre study comparing two different protocols. Thorax . 2012 Feb 29. [QxMD MEDLINE Link] .
Brooks M. FDA OKs Expanded Use of Ivacaftor (Kalydeco) in Cystic Fibrosis. Medscape Medical News. Available at http://www.medscape.com/viewarticle/821097 . Accessed: March 1, 2014.
Gibson RL, Emerson J, McNamara S, et al. Significant microbiological effect of inhaled tobramycin in young children with cystic fibrosis. Am J Respir Crit Care Med . 2003 Mar 15. 167(6):841-9. [QxMD MEDLINE Link] . [Full Text] .
Subbarao P, Stanojevic S, Brown M, et al. Lung clearance index as an outcome measure for clinical trials in young children with cystic fibrosis. A pilot study using inhaled hypertonic saline. Am J Respir Crit Care Med . 2013 Aug 15. 188(4):456-60. [QxMD MEDLINE Link] . [Full Text] .
Milla CE, Ratjen F, Marigowda G, Liu F, Waltz D, Rosenfeld M, et al. Lumacaftor/Ivacaftor in Patients Aged 6-11 Years with Cystic Fibrosis and Homozygous for F508del-CFTR. Am J Respir Crit Care Med . 2017 Apr 1. 195 (7):912-920. [QxMD MEDLINE Link] .
- Chest radiograph of a patient with advanced cystic fibrosis. Note marked hyperinflation, peribronchial thickening, and bilateral infiltrates with evidence of bronchiectasis especially of the upper lobes.
Contributor Information and Disclosures
Girish D Sharma, MD, FCCP, FAAP Professor of Pediatrics, Rush Medical College; Director, Section of Pediatric Pulmonology and Rush Cystic Fibrosis Center, Rush Children's Hospital, Rush University Medical Center Girish D Sharma, MD, FCCP, FAAP is a member of the following medical societies: American Academy of Pediatrics , American College of Chest Physicians , American Thoracic Society , Royal College of Physicians of Ireland Disclosure: Nothing to disclose.
Mary L Windle, PharmD Adjunct Associate Professor, University of Nebraska Medical Center College of Pharmacy; Editor-in-Chief, Medscape Drug Reference Disclosure: Nothing to disclose.
Charles Callahan, DO Professor, Chief, Department of Pediatrics and Pediatric Pulmonology, Tripler Army Medical Center Charles Callahan, DO is a member of the following medical societies: American Academy of Pediatrics , American College of Chest Physicians , American College of Osteopathic Pediatricians , American Thoracic Society , Association of Military Surgeons of the US , Christian Medical and Dental Associations Disclosure: Nothing to disclose.
Kenan Haver, MD Associate Professor of Pediatrics, Harvard Medical School; Director of Asthma Program, Director of Flexible Bronchoscopy Program, Director of Pulmonary Division Asthma Program, Co-Director of Primary Ciliary Dyskinesia, Co-Leader of Empyema Standardized Clinical Assessment and Management Plans (SCAMP), Boston Children’s Hospital Kenan Haver, MD is a member of the following medical societies: American Academy of Pediatrics , American Thoracic Society Disclosure: Author for: UptoDate.
Susanna A McColley, MD Professor of Pediatrics, Northwestern University, The Feinberg School of Medicine; Director of Cystic Fibrosis Center, Head, Division of Pulmonary Medicine, Children's Memorial Medical Center of Chicago Susanna A McColley, MD is a member of the following medical societies: American Academy of Pediatrics , American College of Chest Physicians , American Sleep Disorders Association, American Thoracic Society Disclosure: Received honoraria from Genentech for speaking and teaching; Received honoraria from Genentech for consulting; Partner received consulting fee from Boston Scientific for consulting; Received honoraria from Gilead for speaking and teaching; Received consulting fee from Caremark for consulting; Received honoraria from Vertex Pharmaceuticals for speaking and teaching.
What would you like to print?
- Print this section
- Print the entire contents of
- Print the entire contents of article
- Cystic Fibrosis Imaging
- Sinonasal Manifestations of Cystic Fibrosis
- Surgical Aspects of Cystic Fibrosis and Meconium Ileus
- Pediatric Bronchiectasis
- Bronchiectasis
- Pediatric Bronchitis
- Review Addresses Skin Manifestations of Cystic Fibrosis
- Adults With Cystic Fibrosis Report Poor Sleep, Fatigue, Depression
- Vertex's Triple Combo Cystic-fibrosis Drug Meets Main Late-stage Study Goals

- Drug Interaction Checker
- Pill Identifier
- Calculators

- 2010vanzacaftor-tezacaftor-deutivacaftor-4000472Drugs Drugs vanzacaftor/tezacaftor/deutivacaftor

About Cystic Fibrosis

Cystic fibrosis is a progressive, genetic disease that affects the lungs, pancreas, and other organs.
There are close to 40,000 children and adults living with cystic fibrosis in the United States (and an estimated 105,000 people have been diagnosed with CF across 94 countries), and CF can affect people of every racial and ethnic group.
There are many misconceptions about CF. Learn the facts on our page, Dispelling Misconceptions About Cystic Fibrosis .
In people with CF, mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene cause the CFTR protein to become dysfunctional. When the protein is not working correctly, it’s unable to help move chloride — a component of salt — to the cell surface. Without the chloride to attract water to the cell surface, the mucus in various organs becomes thick and sticky.
In the lungs , the mucus clogs the airways and traps germs, like bacteria, leading to infections , inflammation , respiratory failure, and other complications. For this reason, avoiding germs is a top concern for people with CF.
In the pancreas , the buildup of mucus prevents the release of digestive enzymes that help the body absorb food and key nutrients , resulting in malnutrition and poor growth. In the liver, the thick mucus can block the bile duct, causing liver disease. In men, CF can affect their ability to have children .
Understand how the cystic fibrosis transmembrane conductance regulator (CFTR) affects the GI system.
Today, because of improved medical treatments and care, more than half of people with CF are age 18 or older. Many people with CF can expect to live healthy, fulfilling lives into their 30s, 40s, and beyond.
Read the Foundation's Patient Registry Reports .
People with CF can have a variety of symptoms, including:
- Very salty-tasting skin
- Persistent coughing, at times with phlegm
- Frequent lung infections including pneumonia or bronchitis
- Wheezing or shortness of breath
- Poor growth or weight gain in spite of a good appetite
- Frequent greasy, bulky stools or difficulty with bowel movements
- Nasal polyps
- Chronic sinus infections
- Clubbing or enlargement of the fingertips and toes
- Rectal prolapse
- Male infertility
Learn more about CF — from diagnosis to living with the disease as an adult — in " An Introduction to Cystic Fibrosis: For Patients and Their Families ," or watch the video series.

Jay, a 6-year-old with CF
Listen to CF clinicians explain:
- Which body parts are affected by CF
- Common CF symptoms
- How CF is treated
Cystic fibrosis is a genetic disease . People with CF have inherited two copies of the defective CF gene — one copy from each parent. Both parents must have at least one copy of the defective gene.
People with only one copy of the defective CF gene are called carriers, but they do not have the disease. Each time two CF carriers have a child, the chances are:
- 25 percent (1 in 4) the child will have CF
- 50 percent (1 in 2) the child will be a carrier but will not have CF
- 25 percent (1 in 4) the child will not be a carrier and will not have CF
The defective CF gene contains a slight abnormality called a mutation . There are more than 1,700 known mutations of the disease. Most genetic tests only screen for the most common CF mutations. Therefore, the test results may indicate a person who is a carrier of the CF gene is not a carrier.
Diagnosing cystic fibrosis is a multistep process, and should include a:
- Newborn screening
Genetic or carrier test
- Clinical evaluation at a CF Foundation-accredited care center
Although most people are diagnosed with CF by the age of 2, some are diagnosed as adults. A CF specialist can order a sweat test and recommend additional testing to confirm a CF diagnosis.
Read the CF Foundation’s clinical care guidelines for diagnosing CF .
I grew up wondering why I felt sick every day. As doctors suggested unlikely diseases, such as hormonal disorders, kidney disease, lupus, and depression, I felt I was further from an answer. Then, my ENT suggested CF, a disease I had never heard of. As he described what he knew about CF, it matched all of my symptoms and promised the answer I had been looking for my whole life.” — Katie K., an adult with CF, from the Community Blog
According to the Cystic Fibrosis Foundation Patient Registry, in the United States:
- There are close to 40,000 children and adults living with cystic fibrosis in the United States (and an estimated 105,000 people have been diagnosed with CF across 94 countries).
- Approximately 1,000 new cases of CF are diagnosed each year.
- More than 75 percent of people with CF are diagnosed by age 2.
- More than half of the CF population is age 18 or older.
Did you know?
More than half of the cystic fibrosis population is over 18.
Cystic fibrosis is a complex disease. The types of symptoms and how severe they are can differ widely from person to person. Many different factors can affect a person's health and the course the disease runs, including your age when you are diagnosed.
View this post on Instagram A post shared by Cystic Fibrosis Foundation (@cf_foundation)
The Outlook
Tremendous advancements in specialized CF care have added years and improve the quality of the lives of people with cystic fibrosis. During the 1950s, a child with CF rarely lived long enough to attend elementary school. Today, many people with CF achieving their dreams of attending college , pursuing careers, getting married, and having kids .
Although there has been significant progress in treating this disease, there is still no cure and too many lives are cut far too short.
The types of CF symptoms and how severe they are can differ widely from person to person. Therefore, although treatment plans can contain many of the same elements, they are tailored to each person's unique needs.

Tré, a 24-year-old with CF, wearing his vest.
People with CF and their families have expertise in how the disease affects them and how their daily lives affect the way they approach their care. By acknowledging each other's expertise, people with CF, their families, and clinical care teams can work together to develop treatment plans that align personal life goals with health goals.
“My doctor and I decided to come up with a plan that would work for me. We were able to negotiate a deal so that I was doing more treatments than I had been, but I wasn’t just sitting at home hooked up to machines.” — Betsy Sullivan, a teenager with CF, from the CF Community Blog
Each day, people with CF complete a combination of the following therapies:
- Airway clearance to help loosen and get rid of the thick mucus that can build up in the lungs.
- Inhaled medicines to open the airways or thin the mucus. These are liquid medicines that are made into a mist or aerosol and then inhaled through a nebulizer and include antibiotics to fight lung infections and therapies to help keep the airways clear.
- Pancreatic enzyme supplement capsules to improve the absorption of vital nutrients. These supplements are taken with every meal and most snacks. People with CF also usually take multivitamins.
- An individualized fitness plan to help improve energy, lung function, and overall health
- CFTR modulators to target the underlying defect in the CFTR protein. Because different mutations cause different defects in the protein, the medications that have been developed so far are effective only in people with specific mutations.
Support From the CF Foundation
The CF Foundation supports people with CF by:
- Accrediting more than 130 care centers . These centers are staffed by dedicated health care professionals who provide expert CF care and specialized disease management.
- Supporting research to discover and develop new CF treatments and maintaining a pipeline of potential therapies that target the disease from every angle.
Today, the Foundation is focused on developing lifesaving new therapies for larger numbers of people with CF — including those with rare and nonsense mutations — and pursuing daring, new opportunities to one day develop a lifelong cure.
When a group of parents started the Cystic Fibrosis Foundation in 1955, there were no treatments for cystic fibrosis. These parents set their sights high, to:
- Advance understanding of this little-known disease
- Create new treatments and specialized care for their children
- Find a cure
In the following years, the fundraising and commitment of the CF community has enabled the Foundation to support fundamental research in the laboratory that has led to groundbreaking discoveries , including identifying the gene and protein responsible for cystic fibrosis. By expanding our knowledge of the underlying biology of the disease and its effect on the body, researchers have paved the way for creating new treatments .
The Foundation's steadfast commitment to advancing CF research has helped enable more than a dozen new treatments for the disease. We have made incredible progress, including the approvals by the U.S. Food and Drug Administration (FDA) of Kalydeco ® (ivacaftor), Orkambi ® (lumacaftor/ivacaftor), Symdeko ® (tezacaftor/ivacaftor), Trikafta ® (elexacaftor/tezacaftor/ivacaftor), Cayston ® (aztreonam), and TOBI ® (tobramycin).
Watch this video to see how clinical research has made a difference in the lives of people with CF.
Research by dedicated scientists and clinicians from a wide range of disciplines advances our understanding of cystic fibrosis every day, helping to shape clinical care practices for people living with the disease for years to come. These include studies conducted using patient data in the CF Foundation's Patient Registry , which are helping us identify trends and track the effectiveness of treatments.
The Foundation is supporting the best research here and abroad to improve the quality of life of people with CF today and increase the speed of innovative research and drug development to add tomorrows. Two major initiatives are helping with this mission.
To make meaningful progress against infections , we established the five-year Infection Research Initiative to help improve the detection, diagnosis, prevention, and treatment of infections. From 2018 through 2023, we invested more than $170 million to fund research and the development of new treatments. We also conducted a comprehensive review of our research portfolio, identifying and filling gaps, evaluating our investments, and recalibrating our infection research strategy. This initiative helped us set the agenda going forward for a robust infection research program so that we can continue to meet the needs of the CF community.
The second major initiative is the Path to a Cure , an ambitious research agenda to deliver treatments for the underlying cause of the disease and a cure for every person with CF. The Foundation is challenging potential collaborators to submit proposals that will accelerate the pace of progress in CF drug discovery and development and intends to allocate $500 million to the effort through 2025. The Path to a Cure centers around two core strategies to address the underlying cause of CF: restoring CFTR protein when none exists and fixing or replacing the underlying genetic mutation to address the root cause of CF.
By pursuing these bold strategies and others, the CF Foundation continues to build a robust pipeline of potential new therapies that fight the disease from every angle. Learn more about the CF Foundation's key research programs:
- Research We Fund : See a snapshot of how the CF Foundation is funding cystic fibrosis research.
- CF Foundation Therapeutics Laboratory : Based in Lexington, Mass., the CF Foundation Therapeutics Laboratory identifies and tests potential groundbreaking therapies for CF, readying them for further development.
- Therapeutics Development Network : The Therapeutics Development Network is the largest CF clinical trials network in the world. It provides the resources and support for studies that are leading to important new therapies and better treatments.
- Drug Development Pipeline : Discoveries from the laboratory are being turned into potential drugs that attack both the symptoms of CF and the cause — a faulty gene that makes a defective protein.
- Research Centers : These CF "think tanks" are located at top universities and medical schools across North America, where scientists from many disciplines are brought together to combine their expertise to find a cure for CF.
Cystic Fibrosis
- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE , Medicine , Surgery , Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Table of Contents
Suggest an improvement
- Hidden Post Title
- Hidden Post URL
- Hidden Post ID
- Type of issue * N/A Fix spelling/grammar issue Add or fix a link Add or fix an image Add more detail Improve the quality of the writing Fix a factual error
- Please provide as much detail as possible * You don't need to tell us which article this feedback relates to, as we automatically capture that information for you.
- Your Email (optional) This allows us to get in touch for more details if required.
- Which organ is responsible for pumping blood around the body? * Enter a five letter word in lowercase
- Email This field is for validation purposes and should be left unchanged.
Introduction
Cystic fibrosis (CF) is an inherited disease affecting multiple organs. A genetic mutation results in thickened secretions which commonly leads to recurrent respiratory infections .
CF is the most common inherited disease in Caucasians. 1 In the UK there are around 10,600 cases, whereas worldwide there are thought to be around 100,000. 2
CF is a genetic disease inherited in an autosomal recessive fashion.
Mutations are found on chromosome 7 in the CF transmembrane conductance regulator (CFTR) gene. The most common mutation found in Caucasians is delta-F508 (DF508). 1
Normally, the CFTR allows efflux of chloride and inhibits influx of sodium . Usually this keeps sodium and chloride in the lumen, out of the cells.
In CF, more chloride leaves the cell and more sodium is reabsorbed along with water, due to osmosis. Subsequently, secretions are dehydrated and thickened with increased chloride. 3
Risk factors
As CF is an inherited condition, family history of the disease is the only risk factor. 1
Clinical features
CF can present at any age (although it is usually found in newborn screening) and severity of symptoms varies. 1
CF has effects on multiple systems but most commonly presents with respiratory effects .
Thickened secretions reduce mucociliary clearance from the bronchi and increased salt concentration leads to impaired bacterial defences. Subsequently, bacterial colonisation and lung inflammation increases, which can present as recurrent lower respiratory tract infections and bronchiectasis . 1
Thickened secretions in sinuses can also lead to recurrent infection and subsequent nasal polyps due to chronic inflammation. 4
The gastrointestinal system is affected in several ways. Commonly, pancreatic insufficiency leads to both exocrine and endocrine dysfunction.
Impaired exocrine function leads to malabsorption of nutrients and fat-soluble vitamins, whereas impaired endocrine function results in cystic fibrosis-related diabetes mellitus (CFRD). Blockage of pancreatic ducts can also result in pancreatitis . CF causes thickened bile leading to obstruction of the bile ducts and gallstones . 4
Intestinal obstruction in the newborn, termed meconium ileus , can be the presenting feature in infants. 3 Infants can also present with rectal prolapse. 1 Adults can develop distal intestinal obstruction syndrome (DIOS) which presents with bloating, abdominal pain and vomiting. 4
Infertility is common in boys due to congenital absence or fibrosis of the vas deferens. In girls, thickened mucus in the cervix and irregular menstruation can lead to reduced fertility. 1
Chloride is not resorbed from sweat glands leading to excess salt in the sweat . This allows diagnosis of CF by sweat testing. 1
The history should focus on respiratory symptoms but should also include questions about the less common features of CF:
- Respiratory : frequency of infections, cough, sputum (colour, amount), haemoptysis, breathlessness
- Pancreatic function: failure to thrive, steatorrhoea, thirst, polyuria
- Gastrointestinal : abdominal pain, bloating, vomiting
- Difficulty conceiving and irregular menstruation
Clinical examination
A thorough respiratory examination should be performed. Typical findings on respiratory examination may include:
- General inspection of the environment : sputum pots, oxygen (concentrator or cylinder with a mask or nasal cannulae), nebuliser, inhaler
- General inspection of the patient : younger age, increased work of breathing, cachexia, vascular access (Portacath, PICC line)
- Peripheral signs : finger clubbing (Figure 1), cyanosis
- Chest signs : coarse crackles over areas of bronchiectasis
Examination for other signs of CF should look for:
- Finger prick marks due to insulin use in diabetes
- Abdominal examination for pain, signs of chronic liver disease, feeding tubes (NG or PEG)

Differential diagnoses
Common pathologies such as pneumonia, malignancy and pulmonary embolism should be considered depending on presenting features. If gastrointestinal features are predominant, causes such as coeliac disease should be considered.
A rare but important differential diagnosis is primary ciliary dyskinesia (PCD). This is an autosomal recessive inherited disease caused by dysfunction of cilia in sinuses, lungs and reproductive organs. 4
Investigations
Bedside investigations.
Relevant bedside investigations include:
- Urine dip: for glucose in case of diabetes
- Lung function testing: an obstructive picture is most common but can be restrictive, mixed or normal
Laboratory investigations
Relevant laboratory investigations include: 1
- Blood tests: FBC and CRP (infection and anaemia), U&Es, fasting glucose, LFTs (biliary or liver effects), vitamin A, C and E levels
- Sputum culture and sensitivity: commonly Haemophilus influenzae , Staphylococcus aureus , Pseudomonas aeruginosa , Burkholderia cepacia
- Sweat testing: high chloride concentration in 98% sensitive
- Genetic testing: for CFTR
- Faecal elastase: for pancreatic insufficiency
Relevant imaging investigations include:
- Chest x-ray: bronchiectasis, hyperinflation
- High-resolution CT chest: bronchial wall thickening, bronchiectasis (tree-in-bud appearance, signet ring sign), mucus plugging (finger in glove)
- CT angiography: in haemoptysis, if endovascular intervention is being considered

CF is usually managed by a multidisciplinary team of specialists in a tertiary centre . 1
Medical management
All patients should have annual vaccinations and lifestyle advice for regular physical exercise .
Respiratory manifestations are managed with:
- Chest physiotherapy for mucus clearance with active cycle of breathing
- Prophylactic antibiotics and antibiotics for acute exacerbations guided by sputum culture (oral, inhaled or intravenous)
- Inhaled bronchodilators
- Mucolytics (oral or inhaled): hypertonic saline or dornase alfa
Pancreatic insufficiency is managed with pancreatic enzymes (Creon) and diabetic control (usually with insulin).
Due to recurrent infection and malabsorption, weight should be closely monitored, and nutrition managed by a dietician. Supplementation of fat-soluble vitamins and a high-calorie diet is encouraged. If nutrition cannot be maintained, enteral feeding by PEG or nasogastric tube may be needed.
Fertility counselling should be offered, including information about infertility and the inherent risk in pregnancy.
Follow up should be carried out with specialists twice a year, in conjunction with primary care. It should include regular pulmonary function tests, chest x-ray, sputum cultures and diabetes screening. 1
Surgical management
The mainstay of surgical management is transplant (lung +/- heart). Complications such as massive haemoptysis or pneumothorax may also be managed surgically.
Complications
Respiratory complications are caused by bronchiectasis and recurrent infections. Progressive airflow obstruction can lead to cor pulmonale and death .
Other respiratory complications include haemoptysis (which can be large volume and life-threatening) and pneumothorax .
Other complications include chronic liver disease (and related sequelae), osteoporosis and psychological effects .
- Cystic fibrosis (CF) is an autosomal recessive inherited condition due to a mutation in the CFTR gene . The mutation leads to thickened secretions in multiple organs.
- Thickened mucus in the lungs leads to recurrent infections and bronchiectasis, haemoptysis and pneumothoraces.
- Other effects of thickened secretions include nasal polyps, pancreatic insufficiency (malabsorption and cystic fibrosis-related diabetes mellitus), biliary or intestinal obstruction, infertility and excess salt in sweat.
- History and examination should focus on the respiratory system with a brief abdominal examination. Inspection of the environment and patient is important in CF.
- Differential diagnoses include pneumonia, malignancy, pulmonary embolism and rarely primary ciliary dyskinesia.
- Investigations include lung function testing, basic blood tests, sputum culture, sweat test and genetic testing.
- Appropriate imaging for CF includes chest x-ray and high-resolution CT chest which show bronchiectasis.
- Management of CF is multidisciplinary including chest physiotherapy, antibiotics and mucolytics for respiratory manifestations. Nutritional needs should be managed with a high-calorie diet or enteral feeding, vitamin supplementation and pancreatic enzymes.
- Surgical management can include transplant (heart and/or lung) in some cases.
- Respiratory complications include cor pulmonale, haemoptysis and pneumothorax.
Dr Samantha Cockburn
Respiratory Registrar
Dr Chris Jefferies
- Patient info. Cystic Fibrosis . Published in 2020. Available from: [ LINK ]
- Cystic Fibrosis Trust. Cystic fibrosis FAQs . Available from: [ LINK ]
- Radiopaedia. Cystic fibrosis (pulmonary manifestations). Published in 2010. Available from: [ LINK ]
- National Organization for Rare Disorders. Cystic Fibrosis . Published in 2017. Available from: [ LINK ]
- Radiopaedia/Wikipedia. Finger clubbing in cystic fibrosis. Licence: [ CC BY NC SA 3.0 ]
- Radiopaedia/Dr Jeremy Jones. Bronchiectasis, complicating left sided pneumothorax with chest drain, surgical emphysema, portacath . Licence: [ CC BY NC SA 3.0 ]

Other pages
- Product Bundles 🎉
- Join the Team 🙌
- Institutional Licence 📚
- OSCE Station Creator Tool 🩺
- Create and Share Flashcards 🗂️
- OSCE Group Chat 💬
- Newsletter 📰
- Advertise With Us
Join the community
Overview - Cystic fibrosis
Cystic fibrosis is an inherited condition that causes sticky mucus to build up in the lungs and digestive system. This causes lung infections and problems with digesting food.
In the UK, most cases of cystic fibrosis are picked up at birth using the newborn screening heel prick test.
Symptoms usually start in early childhood and vary from child to child, but the condition gets slowly worse over time, with the lungs and digestive system becoming increasingly damaged.
Treatments are available to help reduce the problems caused by the condition and make it easier to live with, but sadly life expectancy is shortened.
Symptoms of cystic fibrosis
The build-up of sticky mucus in the lungs can cause breathing problems and increases the risk of lung infections. Over time, the lungs may stop working properly.
Mucus also clogs the pancreas (the organ that helps with digestion), which stops enzymes reaching food in the gut and helping with digestion.
This means most people with cystic fibrosis don't absorb nutrients from food properly and need to eat more calories to avoid malnutrition .
Symptoms of cystic fibrosis include:
- recurring chest infections
- wheezing, coughing , shortness of breath and damage to the airways (bronchiectasis)
- difficulty putting on weight and growing
- yellowing of the skin and the whites of the eyes ( jaundice )
- diarrhoea , constipation , or large, smelly poo
- a bowel obstruction in newborn babies (meconium ileus) – surgery may be needed
People with the condition can also develop a number of related conditions, including diabetes , thin, weakened bones (osteoporosis) , infertility in males, and liver problems.
Diagnosing cystic fibrosis
In the UK, all newborn babies are screened for cystic fibrosis as part of the newborn blood spot test (heel prick test) carried out shortly after they're born.
If the screening test suggests a child may have cystic fibrosis, they'll need these additional tests to confirm they have the condition:
- a sweat test – to measure the amount of salt in sweat, which will be abnormally high in someone with cystic fibrosis
- a genetic test – where a sample of blood or saliva is checked for the faulty gene that causes cystic fibrosis
These tests can also be used to diagnose cystic fibrosis in older children and adults who didn't have the newborn test.
The genetic test can also be used to see whether someone is a "carrier" of cystic fibrosis in cases where the condition runs in the family.
This test can be important for someone who thinks they may have the faulty gene and wishes to have children.
The Cystic Fibrosis Trust has more information about genetic testing for cystic fibrosis.
Treatments for cystic fibrosis
There's no cure for cystic fibrosis, but a range of treatments can help control the symptoms, prevent or reduce complications, and make the condition easier to live with.
People with cystic fibrosis may need to take different medicines to treat and prevent lung problems.
Physical activity and the use of airway clearance techniques may also be recommended to help clear mucus from the lungs.
Find out more about treatments for cystic fibrosis .
Complications of cystic fibrosis
People with cystic fibrosis also have a higher risk of developing other conditions.
These include:
- weak and brittle bones (osteoporosis) – medicines called bisphosphonates can sometimes help
- diabetes – insulin and a special diet may be needed to control blood sugar levels
- nasal polyps and sinus infections – steroids, antihistamines, antibiotics or sinus flushes can help
- liver problems
- fertility problems – it's possible for women with cystic fibrosis to have children, but men won't be able to father a child without help from fertility specialists (see a doctor or fertility specialist for more advice)
People with cystic fibrosis should not meet face to face. This is because they're more likely to spread infections, and more vulnerable to complications if they do develop an infection.
The Cystic Fibrosis Trust has more information about complications of cystic fibrosis and preventing cross-infection .
Cause of cystic fibrosis
Cystic fibrosis is a genetic condition. It's caused by a faulty gene that affects the movement of salt and water in and out of cells.
This, along with recurrent infections, can result in a build-up of thick, sticky mucus in the body's tubes and passageways – particularly the lungs and digestive system.
A person with cystic fibrosis is born with the condition. It's not possible to "catch" cystic fibrosis from someone else who has it.
How cystic fibrosis is inherited
To be born with cystic fibrosis, a child has to inherit a copy of the faulty gene from both of their parents.
This can happen if the parents are "carriers" of the faulty gene, which means they don't have cystic fibrosis themselves.
It's estimated around 1 in every 25 people in the UK are carriers of cystic fibrosis.
If both parents are carriers, there's a:
- 1 in 4 chance their child won't inherit any faulty genes and won't have cystic fibrosis or be able to pass it on
- 1 in 2 chance their child will inherit a faulty gene from one parent and be a carrier
- 1 in 4 chance their child will inherit the faulty gene from both parents and have cystic fibrosis
If one parent has cystic fibrosis and the other is a carrier, there's a:
- 1 in 2 chance their child will be a carrier
- 1 in 2 chance their child will have cystic fibrosis
Cystic fibrosis tends to get worse over time and can be fatal if it leads to a serious infection or the lungs stop working properly.
But people with cystic fibrosis are now living for longer because of advancements in treatment.
Currently, about half of people with cystic fibrosis will live past the age of 40. Children born with the condition nowadays are likely to live longer than this.
Support is available to help people with cystic fibrosis live as independently as they can and have the best possible quality of life.
It can be helpful to speak to others who have the same condition, and to connect with a charity.
The following links may be useful:
- Asthma + Lung UK – the UK's lung health charity
- Cystic Fibrosis Trust – which has an online forum and news about ongoing research into cystic fibrosis
- CF Kids – a support group for parents of children with cystic fibrosis
- Cystic Fibrosis Care – a charity that provides practical help and support
Information about you
If you or your child has cystic fibrosis, your clinical team will ask you if you consent to being on the UK Cystic Fibrosis Registry.
This is a secure anonymous registry sponsored by the Cystic Fibrosis Trust that records health information on people with cystic fibrosis.
The registry helps scientists look for better ways to prevent and treat this condition. You can opt out of the register at any time.
Find out more about the UK Cystic Fibrosis Registry
Page last reviewed: 16 March 2021 Next review due: 16 March 2024
- Remote Access
- Save figures into PowerPoint
- Download tables as PDFs

Chapter 38: Presentation and Management of Cystic Fibrosis
Blakeslee E. Noyes; Andrew J. Lechner
- Download Chapter PDF
Disclaimer: These citations have been automatically generated based on the information we have and it may not be 100% accurate. Please consult the latest official manual style if you have any questions regarding the format accuracy.
Download citation file:
- Search Book
Jump to a Section
Introduction and historical background, etiology and genetics of cystic fibrosis.
- NEWBORN SCREENING FOR CYSTIC FIBROSIS
- ORGAN SYSTEM MANIFESTATIONS OF CYSTIC FIBROSIS
- TREATMENT AND MONITORING OF CF PULMONARY DISEASE
- LUNG TRANSPLANTATION FOR CYSTIC FIBROSIS
- SURVIVAL IN CYSTIC FIBROSIS
- SUGGESTED READINGS
- Full Chapter
- Supplementary Content
The student will be able to describe the genetics, most common gene mutations, and protein abnormalities that characterize cystic fibrosis (CF).
The student will be able to define the competing hypotheses that have been proposed to cause the clinical manifestations of CF.
The student will be able to outline the approaches to identify CF patients through newborn screening, and describe the typical presenting signs and symptoms of CF lung and gastrointestinal disease.
The student will be able to identify the sequence of events that occurs in CF and the rationale for treatment interventions, including for non-pulmonary organs.
Cystic fibrosis (CF) was first formally described in 1938 by the pathologist Dorothy Andersen, who recognized a number of patients with characteristic lesions of the pancreas and a clinical syndrome of failure to thrive, diarrhea, and recurrent respiratory infections. However, CF was recognized in European folklore hundreds of years earlier, and anticipated the sweat electrolyte abnormalities in CF. An anonymous medieval German source wrote, "Woe is the child who tastes salty from a kiss on the brow, for he is cursed, and soon must die." Shortly after Andersen's description, CF was recognized as an autosomal recessive disorder. By the mid-1950s, sweat electrolyte abnormalities were identified in CF and the technique to perform a sweat test was described that remains the gold standard to this day. Discovery of the CF gene in 1989 provided a critical advance in understanding the basic defect and pathophysiology in CF, and triggered a number of important advances in treating CF patients. As a direct result, the prognosis for patients with CF continues to improve (see below).
Cystic fibrosis is now recognized as the most common lethal genetic disease in the Caucasian population, with an estimated 30,000 patients in the United States and 27,000 in Europe. CF is most common among Caucasians of northern European descent, with a disease prevalence of ~1 in 3,000 births, and the frequency of being a carrier of a defective CF gene is estimated as 1 in 29. The disease occurs less commonly in other ethnic groups, having approximate incidences of 1 in 4,000-10,000 Latin Americans, 1 in 15,000 African Americans, and 1 in 35,000 Asian Americans.
CF is an autosomal recessive disorder caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator ( CFTR ) protein. The CFTR gene is located on the long arm of chromosome 7 at position 7q31. Over 2,500 distinct CFTR mutations have been identified, but deletion of phenylalanine at position 508 ( ΔF508 ) of the CFTR protein is by far the most common. Between 65% and 75% of CF patients in the United States are homozygous or compound heterozygous for a ΔF508 mutation.
Get Free Access Through Your Institution
Pop-up div successfully displayed.
This div only appears when the trigger link is hovered over. Otherwise it is hidden from view.
Please Wait
There are many manifestations of CF that can suggest the diagnosis.
Presentations
Prenatal diagnosis.
Pregnant women may have a simple blood test to look for common mutations (abnormalities) in the CFTR gene that cause cystic fibrosis (CF). If she carries one CFTR gene mutation. There is a 50 percent chance that this CFTR gene mutation will be passed on to the baby. If the father is also a carrier of a CFTR gene mutation there is a 25 percent (1 in 4) chance that the baby will have CF. Amniocentesis or chorionic villus sampling (CVS) sampling can be used for prenatal diagnosis of the baby. Prenatal screening does not test for all the known CFTR gene mutations, therefore, children can still be diagnosed with CF even if prenatal testing was normal or negative.

Inheritance pattern of normal (N) or mutated (M) CFTR genes from parents who carry an abnormal copy of the CFTR gene.
Newborn Screening

All 50 states perform newborn screening of infants to test for hypothyroidism, phenylketonuria, and several other diseases, including CF. The newborn screen tests a small amount of blood, usually obtained from the baby’s heel. The goal of newborn screening is to diagnose CF before any symptoms develop. Newborn screening tests vary from state to state. Maryland added a test for CF to its newborn screening program in 2006. Not all infants with a “positive” newborn screen have CF. Therefore, infants with a “positive” newborn screening test for CF should be referred for a sweat test to confirm the diagnosis.
Additionally, newborn screening does not detect all children with CF. Therefore, children with symptoms that could be caused by CF should be evaluated with a sweat test even if the newborn screening was “normal” or “negative.”
Meconium Illeus
Approximately 18 percent of infants with CF present with meconium ileus (MI), which is an obstruction of the bowel caused by thick, abnormal meconium. Meconium ileus is suspected if a baby fails to pass meconium shortly after birth and develops symptoms of a bowel obstruction, such as abdominal distention or vomiting. Meconium ileus can lead to bowel perforation, a twisting of the bowel, or inflammation and infection in the abdomen.
Meconium ileus must be treated immediately to prevent complications. In some cases, enemas can be used to flush out the meconium. However, in severe cases, surgery is required to remove the obstruction. All babies with meconium ileus should be tested for CF because 98 percent of full-term babies with meconium ileus have CF.
A hyperechoic or echogenic bowel pattern is a prenatal ultrasound finding that suggests an intestinal obstruction caused by abnormal meconium. Among fetuses who have a prenatal ultrasound with hyperechoic bowel, about 1 in 10 will have CF and meconium ileus. In pregnancies where there is evidence of fetal bowel obstruction on prenatal ultrasound, both parents can be tested to find out if they carry CFTR gene mutations. Sometimes an echogenic bowel is a transient finding that resolves by the third trimester.

A. Illustration of intestine blocked by meconium. B. Abdominal x-ray of a newborn infant with meconium ileus showing dilated loops of bowel.
Respiratory Problems

Compare the normal lung airway to the lung airway with CF and bacterial infection or with CF and inflammation.
The respiratory problems associated with CF can present at any age, and can affect both the upper and lower respiratory tracts. Recurrent or chronic sinus infections are common in people with CF. The possibility of CF should be considered in individuals with severe sinus disease or nasal polyps. Sinus CT scans of people with CF almost always show abnormal sinus cavities full of mucus.
Recurrent respiratory symptoms or infections may suggest the diagnosis of CF. The most prominent feature of lower respiratory tract disease in CF is a chronic cough. Sputum production is present in more severe lung disease.
Chest x-rays often show hyperinflation, small collapsed portions of the lung, mucus pugging or infection. People with CF may be affected by recurrent pneumonia, bronchitis and/or wheezing. Bronchiectasis can be observed in CT scans of the chest. Airways infection with certain bacteria such as Pseudomonas aeruginosa also suggest the diagnosis of CF.
Gastrointestinal Problems
Pancreatic insufficiency.
The gastrointestinal tract is frequently affected in CF. Approximately 80 percent of individuals with CF have pancreatic insufficiency. In these people, abnormal secretions block the passages within the pancreas leading to scarring and inadequate enzyme excretion into the intestine. Pancreatic insufficiency leads to malabsorption, (improper digestion of food) especially fats and difficulty gaining weight. In children, poor weight gain often results in growth below the standard growth curves and is called failure to thrive. Children with failure to thrive should be tested for CF, especially if they have abnormal stools or respiratory problems.
| Signs and Symptoms of Malabsorption |
|---|
| Abdominal Bloating |
| Abdominal Pain |
| Cramping |
| Fat soluble vitamin (A, E, D, and K) deficiency |
| Flatulence (excess gas) |
| Frequent bulky, greasy or oily, or foul-smelling stools (Steatorrhea) |
| Malnutrition or poor weight gain |
Unusual Presentations
Cftr-related disorder.
Most individuals with CF have respiratory and gastrointestinal problems. However, some people without the usual features of CF may have a “positive” (abnormal) or intermediate sweat test . They may have only one or no known CFTR gene mutations, instead of the two mutations seen in most people with CF. The diagnosis of CFTR-related disorder is applied to these people with these atypical presentations. Symptoms of CFTR-related disorder may include chronic sinus disease, nasal polyps, pancreatitis, or males infertility. Individuals with atypical CF presentations usually do not have pancreatic insufficiency and they are often diagnosed at an older age.
Chronic Sinus Disease
The sinuses are affected in almost all individuals with CF. CT scans of the sinuses almost always show abnormalities including sinuses filled with mucus or pus rather than air. Recurrent sinus infections can be a sign of CF, especially if caused by certain bacteria such as Pseudomonas aeruginosa .
The mucosa, or lining, of the nasal passages in people with CF is often red and swollen. Nasal polyps may develop in this inflamed nasal mucosa. Symptoms of nasal polyps include stuffy nose, mouth breathing, snoring, nasal pain, change in voice, distortion or widening of the nasal bridge, or nose bleeds. Because nasal polyps are uncommon in the general population, individuals with nasal polyps should be tested for CF.
Pancreatitis
Acute pancreatitis has been reported in about 15 percent of individuals with CF. Symptoms include severe abdominal pain and vomiting. People with pancreatitis may or may not have chronic pulmonary symptoms. Individuals with pancreatitis typically produce adequate pancreatic enzymes and are not affected by malabsorption or failure to thrive.
Congenital Bilateral Absence of the Vas Deferens (CBAVD)
Congenital bilateral absence of the vas deferens (CBAVD) leads to the absence of sperm (azoospermia) in men with CF. Almost all post-pubertal males with CF have azoospermia, leading to infertility. Some men with CBAVD due to CFTR gene mutations have no other features of CF. These individuals may have normal, intermediate, or elevated sweat chloride concentrations. People with CBAVD should be monitored for the development of other CF-related complications.
Log in using your username and password
- Search More Search for this keyword Advanced search
- Latest content
- Current issue
- BMJ Journals
You are here
- Volume 90, Issue 1
- Diagnosis and management of cystic fibrosis
- Article Text
- Article info
- Citation Tools
- Rapid Responses
- Article metrics
- Rosalind L Smyth
- For correspondence: Professor Rosalind L Smyth Division of Child Health, School of Reproductive and Developmental Medicine, University of Liverpool Institute of Child Health, Alder Hey Children’s Hospital, Liverpool L12 2AP, UK; r.l.smythliv.ac.uk
https://doi.org/10.1136/adc.2005.074021
Statistics from Altmetric.com
Request permissions.
If you wish to reuse any or all of this article please use the link below which will take you to the Copyright Clearance Center’s RightsLink service. You will be able to get a quick price and instant permission to reuse the content in many different ways.
- cystic fibrosis
Cystic fibrosis (CF) is the most common life limiting genetic disorder of white populations. There are over 6000 patients in the UK with this condition and at least 30 000 in the USA. 1 This review focuses on the challenges of diagnosis, clinical complications, and management of children with CF.
DIAGNOSIS OF CF
- In this window
- In a new window
Presentation of cystic fibrosis (CF)
Sweat chloride > 60 mmol/l is considered diagnostic of CF. Values of sweat chloride of 40–60 mmol/l are considered highly suggestive of CF, and in this situation the sweat test should be repeated and/or genetic testing conducted. Although in CF patients there is a fall in sweat chloride with age, the magnitude of this fall is not sufficient to cause diagnostic confusion, if these criteria are used. 2 Sweat sodium measurements are less reliable as concentrations of 60–80 …
Read the full text or download the PDF:
Cystic Fibrosis
- First Online: 19 July 2024
Cite this chapter

- Christine M. Bojanowski 6 ,
- Ross C. Klingsberg 6 , 7 &
- Michael Landry 7
With advances in medical care and with the introduction of highly effective modulator therapies, the predicted median survival of individuals with cystic fibrosis has increased dramatically over the last several decades. As a result, over half of the current cystic fibrosis population is 18 years of age or older, making it likely that many primary care providers will see patients with cystic fibrosis in their adult medical practices. With expanded knowledge of cystic fibrosis genetics and genotype:phenotype relationships, it is also possible that primary care providers will be able to recognize and diagnose previously missed adults with cystic fibrosis. Though the comprehensive care of patients with cystic fibrosis is best directed by one of the nationwide Cystic Fibrosis Foundation-accredited care centers, primary care providers will need to play an increasingly critical role in the long-term co-management of adult patients with cystic fibrosis. The purpose of this chapter is to educate the primary care provider about common medical problems experienced by patients with cystic fibrosis, to provide some fundamental knowledge of the cornerstones of chronic cystic fibrosis disease management, and to highlight key issues when co-management with subspecialists with expertise in the field of cystic fibrosis is indicated. Key concepts in care management for cystic fibrosis patients transitioning from pediatric- to adult-oriented healthcare systems will also be reviewed.
This is a preview of subscription content, log in via an institution to check access.
Access this chapter
Subscribe and save.
- Get 10 units per month
- Download Article/Chapter or eBook
- 1 Unit = 1 Article or 1 Chapter
- Cancel anytime
- Available as PDF
- Read on any device
- Instant download
- Own it forever
- Available as EPUB and PDF
- Compact, lightweight edition
- Dispatched in 3 to 5 business days
- Free shipping worldwide - see info
Tax calculation will be finalised at checkout
Purchases are for personal use only
Institutional subscriptions
Cystic Fibrosis Foundation. Cystic Fibrosis Patient Registry Annual Data Report. 2020. https://www.cff.org/medical-professionals/patient-registry
United States Cystic Fibrosis Foundation, Johns Hopkins University, the hospital for sick children. Clinical and functional translation of CFTR. 2011. www.cftr2.org .
Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: cystic fibrosis foundation consensus report. J Pediatr. 2008;153(2):S4–14.
Article PubMed PubMed Central Google Scholar
Moran A, Dunitz J, Nathan B, Saeed A, Lolme B, Thomas W. Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care. 2009;32(9):1626–31.
Moran A, Brunzell C, Cohen RC, Katz M, Marshall BC, Onady G, et al. Clinical care guidelines for cystic fibrosis-related diabetes. Diabetes Care. 2010;33(12):2697–708.
Flume PA, Mogayzel PJ, Robinson KA, Rosenblatt RL, Quittell L, Marshall BC. Cystic fibrosis pulmonary guidelines. Pulmonary complications: hemoptysis and pneumothorax. Am J Respir Crit Care Med. 2010;182(3):298–306.
Article PubMed Google Scholar
Anderson DH. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. Am J Dis Child. 1938;56(2):344–99.
Article Google Scholar
Tobias E, Connor M, Ferguson-Smith M. Ch 15. Strong family history—typical Mendelian disease. In: Tobias ES, editor. Essential medical genetics, 6E. Hoboken, NJ: Wiley; 2011. p. 312.
Google Scholar
MacKenzie T, Gifford AH, Sabadosa KA, Quinton HB, Knapp EA, Goss CH, Marshall BC. Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the cystic fibrosis foundation patient registry. Ann Intern Med. 2014;161(4):233–41.
Beall RJ. The cystic fibrosis foundation. In: Yankaskas JR, Knowles MR, editors. Cystic fibrosis in adults. Riverwoods, IL: Walters Kluwer Publishers; 1999. p. 477–84.
Boyle MP. Adult cystic fibrosis. JAMA. 2007;298(15):1787–93.
Article CAS PubMed Google Scholar
Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med. 2006;354(3):241–50.
Chaudary N, Balasa G. Airway clearance therapy in cystic fibrosis patients insights from a clinician providing cystic fibrosis care. Int J Gen Med. 2021;14:2513–21.
Flume PA, Robinson KA, O’Sullivan BP, Finder JD, Vender RL, Willey-Courand DB, et al. Cystic fibrosis pulmonary guidelines: airway clearance therapies. Respir Care. 2009;54(4):522–37.
PubMed Google Scholar
Aris RM, Merkel PA, Bachrach LK, Borowitz DS, Boyle MP, Elkin SL, et al. Guide to bone health and disease in cystic fibrosis. J Clin Endocrinol Metab. 2005;90(3):1888–96.
Quittner AL, Abbott J, Georgiopoulos AM, Goldbeck L, Smith B, Hempstead SE, et al. International committee on mental health in cystic fibrosis: Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus statements for screening and treating depression and anxiety. Thorax. 2016;71(1):26–34.
Popli K, Stewart J. Infertility and its management in men with cystic fibrosis: review of literature and clinical practices in the UK. Hum Fertil (Camb). 2007;10(4):217–21.
Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D. Cystic fibrosis adult care. Chest. 2004;125(1 Suppl):1S–39S.
Rousset-Jablonski C, Reynaud Q, Nove-Josserand R, Ray-Coquard I, Mekki Y, Golfier F, Durieu I. High proportion of abnormal pap smear tests and cervical dysplasia in women with cystic fibrosis. Eur J Obstet Gynecol Reprod Biol. 2018;221:40–5.
Thornton C, somayaji R, Chu A, Parkins M. Human papillomavirus (HPV) and cervical dysplasia in adult female cystic fibrosis (CF) lung transplant recipients. Thora. 2022;77(6):625–7. https://doi.org/10.1136/thoraxjnl-2021-218461 .
Okumur MJ, Kleinhenz ME. Cystic fibrosis transitions of care: lessons learned and future directions for cystic fibrosis. Clin Chest Med. 2016;37(1):119–26.
Download references
Author information
Authors and affiliations.
Department of Medicine, Tulane University School of Medicine, New Orleans, LA, USA
Christine M. Bojanowski & Ross C. Klingsberg
Department of Internal Medicine, Southeast Louisiana Veterans Healthcare System and Tulane University School of Medicine, New Orleans, LA, USA
Ross C. Klingsberg & Michael Landry
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Ross C. Klingsberg .
Editor information
Editors and affiliations.
Medicine-Pediatrics, Univ of California, Los Angeles, Los Angeles, CA, USA
Alice A Kuo
Medicine and Pediatrics, Northwell Health, Lake Success, NY, USA
Mariecel Pilapil
Medicine, University of Cincinnati College of Medicine, Cincinnati, OH, USA
David E. DeLaet
Dept of Medicine and Pediatrics, Baylor College of Medicine, Houston, TX, USA
Cynthia Peacock
Medicine, Pediatrics, Brigham and Women's Hospital/Boston Children’s Hospital, Boston, MA, USA
Niraj Sharma
Cystic fibrosis (CF) fact sheet | |
|---|---|
Definition and symptoms |
• Inherited abnormalities in the cystic fibrosis transmembrane conductance regulator (CFTR), an epithelial cell membrane protein and chloride channel • Abnormal CFTR protein disrupts chloride transport and water movement across secretory epithelial membranes • Thick (viscous) mucus buildup in the lungs, pancreas, and other organs • Bronchiectasis (persistent lung infections and progressive obstructive airway disease) • Persistent coughing, at times with phlegm • Hemoptysis and pneumothorax • Exocrine pancreatic enzyme insufficiency with poor growth or weight gain in spite of good appetite • Frequent greasy, bulky stools, or difficulty with bowel movements • Male (not female) infertility |
Prevalence | In the United States • About 31,000 people are living with cystic fibrosis (70,000 worldwide) • Approximately 1000 new cases of CF are diagnosed each year • More than 75% of people with CF are diagnosed by age 2 years • Over half of the CF population is aged 18 years or older • Predicted median survival: 50 years |
Genetics and epidemiology | Epidemiology • CF is the most common disorder of autosomal recessive inheritance in Caucasians • In general, it occurs in 1/3000 Caucasian, 1/6000 Hispanic, 1/10,000 African-American, and 1/90,000 Asian American births Genetics: • More than 2100 mutations in the CFTR gene have been identified • The most common CF-causing mutation is Phe508del • The most common CF-causing genes are screened for at birth • People with only one copy of the defective CF gene are called carriers, but they do not have the disease. Each time two CF carriers have a child; the chances are as follows: – 25% (1 in 4) of the offspring will have CF – 50% (1 in 2) of the offspring will be a carrier but will not have CF – 25% (1 in 4) of the offspring will not be a carrier and will not have CF |
Characteristics of adults with CF 18 years and older | • 8.4% masters/doctoral-level degree • 30.7% college graduate • 29.3% some college • 24.1% high school diploma • 44.9% married/co-habitating • 40.0% full-time employment • 99.1% have medical insurance • Phe508del homozygotes—44.5% of CF population • Phe508del heterozygotes—41.2% of CF population |
Associated conditions | Individuals with CF have high rates of comorbid physical and mental health conditions, including • Bronchiectasis • Airway colonization and chronic infections with , , and spp. • Allergic bronchopulmonary aspergillosis • Nontuberculous mycobacterial infection • Exocrine pancreatic insufficiency • CF-related diabetes • Fat-soluble vitamin (A, D, E, and K) deficiency • Osteopenia/osteoporosis • Male infertility • Hepatobiliary disease (cirrhosis, gallstones, biliary disease) • Pancreatitis • Intestinal obstruction, intussusception • Chronic pain • Depression, anxiety, substance abuse, and suicide |
Challenges in transition | • Learning to manage time, finances, housing, employment, education, medication, and frequent physician visits • Challenges impacting the transition to adulthood include – Parental educational and financial status – Proximity to specialized CF care centers – Financial burden of medications and inability to work due to illness – Unpredictable nature of exacerbations requiring intensification of care and usually IV antibiotics and disruption of usual activities – Reluctance on the part of patient and parents/caregivers and pediatric providers to initiate transition – Difficulty identifying adult generalist willing to accept patient into practice |
- Adapted from Cystic Fibrosis Patient Registry Annual Data Report 2020 [ 1 ]
Rights and permissions
Reprints and permissions
Copyright information
© 2024 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter
Bojanowski, C.M., Klingsberg, R.C., Landry, M. (2024). Cystic Fibrosis. In: Kuo, A.A., Pilapil, M., DeLaet, D.E., Peacock, C., Sharma, N. (eds) Care of Adults with Chronic Childhood Conditions. Springer, Cham. https://doi.org/10.1007/978-3-031-54281-7_28
Download citation
DOI : https://doi.org/10.1007/978-3-031-54281-7_28
Published : 19 July 2024
Publisher Name : Springer, Cham
Print ISBN : 978-3-031-54280-0
Online ISBN : 978-3-031-54281-7
eBook Packages : Medicine Medicine (R0)
Share this chapter
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Publish with us
Policies and ethics
- Find a journal
- Track your research

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
- My Bibliography
- Collections
- Citation manager
Save citation to file
Email citation, add to collections.
- Create a new collection
- Add to an existing collection
Add to My Bibliography
Your saved search, create a file for external citation management software, your rss feed.
- Search in PubMed
- Search in NLM Catalog
- Add to Search
Caregivers' perspectives on decision making about lung transplantation in cystic fibrosis
Affiliation.
- 1 Department of Pediatrics, Division of Pulmonology, CB #7217, 5119-B Bioinformatics Building, 130 Mason Farm Road, UNC School of Medicine, Chapel Hill, NC 27599-7217, USA. [email protected]
- PMID: 20050454
- PMCID: PMC3757089
- DOI: 10.1177/152692480901900406
Context: Lung transplantation extends survival for some patients with advanced cystic fibrosis, but it is complicated, has many potential risks, and its outcomes are difficult to predict. No standards exist for informed decision making about transplantation.
Objective: To assess decision making from the perspective of caregivers of patients who faced the transplant decision before dying of cystic fibrosis or transplant complications.
Design: Semistructured interviews with descriptive and qualitative content analysis.
Participants: Twenty-eight caregivers of patients with cystic fibrosis who received care at our center and died between 1996 and 2006.
Results: Of 28 patients who considered lung transplantation, 19 (68%) received transplants, 6 (21%) died while waiting for transplant, and 3 (11%) declined transplant. Three caregivers (11%) thought that the patient did not fully understand the reason for transplant referral. Five (18%) thought that the patient did not fully understand potential risks. Ten (36%) thought that alternatives were not fully understood. The only alternatives to transplant identified, progressive illness and the possibility of earlier death without transplant, were unacceptable to most. Thirteen caregivers (46%) reported that the patient thought that declining transplant was not an option. Caregivers described the decision as "easy" for 19 (68%), often expressing a sentiment of "do or die." Those who described the decision as "easy" recalled fewer elements of informed decision making.
Conclusions: From caregivers' reports, patients with cystic fibrosis may not fully understand risks of and alternatives to lung transplantation. Because a strong desire to prolong life necessitates honest communication about potential outcomes, interventions are needed to facilitate high-quality decision making.
PubMed Disclaimer
Similar articles
- Qualitative Analysis of Perspectives on Lung Transplant among People with Cystic Fibrosis. Milinic T, Hobler MR, Bartlett LE, Gill E, Burdis N, Engelberg RA, Curtis JR, Hartzler AL, Aitken ML, Kapnadak SG, Goss CH, Smith PJ, Ramos KJ. Milinic T, et al. Ann Am Thorac Soc. 2024 Jul;21(7):1044-1052. doi: 10.1513/AnnalsATS.202307-625OC. Ann Am Thorac Soc. 2024. PMID: 38259137
- Development of a decision aid for adult cystic fibrosis patients considering referral for lung transplantation. Vandemheen KL, Aaron SD, Poirier C, Tullis E, O'Connor A. Vandemheen KL, et al. Prog Transplant. 2010 Mar;20(1):81-7. doi: 10.1177/152692481002000113. Prog Transplant. 2010. PMID: 20397351
- Implementation of a cystic fibrosis lung transplant referral patient decision aid in routine clinical practice: an observational study. Stacey D, Vandemheen KL, Hennessey R, Gooyers T, Gaudet E, Mallick R, Salgado J, Freitag A, Berthiaume Y, Brown N, Aaron SD. Stacey D, et al. Implement Sci. 2015 Feb 7;10:17. doi: 10.1186/s13012-015-0206-4. Implement Sci. 2015. PMID: 25757139 Free PMC article.
- Lung transplant referral for individuals with cystic fibrosis: Cystic Fibrosis Foundation consensus guidelines. Ramos KJ, Smith PJ, McKone EF, Pilewski JM, Lucy A, Hempstead SE, Tallarico E, Faro A, Rosenbluth DB, Gray AL, Dunitz JM; CF Lung Transplant Referral Guidelines Committee. Ramos KJ, et al. J Cyst Fibros. 2019 May;18(3):321-333. doi: 10.1016/j.jcf.2019.03.002. Epub 2019 Mar 27. J Cyst Fibros. 2019. PMID: 30926322 Free PMC article. Review.
- Selection of cystic fibrosis patients for lung transplantation. Aurora P, Carby M, Sweet S. Aurora P, et al. Curr Opin Pulm Med. 2008 Nov;14(6):589-94. doi: 10.1097/MCP.0b013e328313e3d4. Curr Opin Pulm Med. 2008. PMID: 18812837 Review.
- Patient Experiences and Perspectives of Their Decision-Making to Accept Lung Transplantation Referral: A Qualitative Study. Chen M, Zou X, Nan J, Nuerdawulieti B, Huxitaer X, Jiang Y. Chen M, et al. Int J Environ Res Public Health. 2023 Mar 5;20(5):4599. doi: 10.3390/ijerph20054599. Int J Environ Res Public Health. 2023. PMID: 36901608 Free PMC article.
- Models of Palliative Care Delivery for Individuals with Cystic Fibrosis: Cystic Fibrosis Foundation Evidence-Informed Consensus Guidelines. Kavalieratos D, Georgiopoulos AM, Dhingra L, Basile MJ, Rabinowitz E, Hempstead SE, Faro A, Dellon EP. Kavalieratos D, et al. J Palliat Med. 2021 Jan;24(1):18-30. doi: 10.1089/jpm.2020.0311. Epub 2020 Sep 16. J Palliat Med. 2021. PMID: 32936045 Free PMC article.
- Choosing between medical management and liver transplant in urea cycle disorders: A conceptual framework for parental treatment decision-making in rare disease. Gerstein MT, Markus AR, Gianattasio KZ, Le Mons C, Bartos J, Stevens DM, Mew NA. Gerstein MT, et al. J Inherit Metab Dis. 2020 May;43(3):438-458. doi: 10.1002/jimd.12209. Epub 2020 Jan 13. J Inherit Metab Dis. 2020. PMID: 31883128 Free PMC article.
- Prevalence of unmet palliative care needs in adults with cystic fibrosis. Trandel ET, Pilewski JM, Dellon EP, Jeong K, Yabes JG, Moreines LT, Arnold RM, Hoydich ZP, Kavalieratos D. Trandel ET, et al. J Cyst Fibros. 2020 May;19(3):394-401. doi: 10.1016/j.jcf.2019.11.010. Epub 2019 Dec 18. J Cyst Fibros. 2020. PMID: 31862306 Free PMC article.
- Atlas is not alone: sharing the burden of clinical challenge. Bribriesco A, Raja S, Ahmad U. Bribriesco A, et al. J Thorac Dis. 2018 Apr;10(Suppl 9):S972-S973. doi: 10.21037/jtd.2018.03.78. J Thorac Dis. 2018. PMID: 29850178 Free PMC article. No abstract available.
- Davis PB, di Sant’Agnese PA. Assisted ventilation for patients with cystic fibrosis. JAMA. 1978;239(18):1851–1854. - PubMed
- [Accessed May 8, 2009];Cystic Fibrosis Foundation Patient Registry: Annual Data Report. 2007 http://www.cff.org/research/ClinicalResearch/PatientRegistryReport .
- Egan TM, Murray S, Bustami RT, et al. Development of the new lung allocation system in the United States. Am J Trans-plant. 2006;6(5 pt 2):1212–1227. - PubMed
- Gee L, Abbott J, Hart A, Conway SP, Etherington C, Webb AK. Associations between clinical variables and quality of life in adults with cystic fibrosis. J Cyst Fibros. 2005;4(1):59–66. - PubMed
- Yankaskas JR, Knowles MR, editors. Clinical Pathophysiology and Manifestations of Lung Disease: Cystic Fibrosis in Adults. Lippincott-Raven; Philadelphia, PA: 1999.
Publication types
- Search in MeSH
Related information
- Cited in Books
Grants and funding
- R01 NR009826/NR/NINR NIH HHS/United States
- U18 HS010397/HS/AHRQ HHS/United States
- U18HS10397-06/HS/AHRQ HHS/United States
LinkOut - more resources
Full text sources.
- Europe PubMed Central
- PubMed Central
- Genetic Alliance
- MedlinePlus Health Information

- Citation Manager
NCBI Literature Resources
MeSH PMC Bookshelf Disclaimer
The PubMed wordmark and PubMed logo are registered trademarks of the U.S. Department of Health and Human Services (HHS). Unauthorized use of these marks is strictly prohibited.

Adventures in camping outdoors with a child who has cystic fibrosis
I've learned to make preparations while also expecting the unexpected

by Jennifer Chamberlain | July 16, 2024
Share this article:

As the dust swirled nearby, I adjusted my daughter’s mask. We were in the middle of nowhere, off-roading to a mining ghost town with no cell service. The scenery was gorgeous, yet I began to second guess my decision. Was I putting her at risk for a day of adventure? What would happen if an emergency happened so far from help?
The great outdoors can pose many risks for a person with cystic fibrosis (CF). Pseudomonas aeruginosa , for instance, is a common and harmful bacteria for those living with the disease . It’s commonly found in water and soil, two large features of camping.
Yet despite these risks, the past two summers our family has spent seven weeks — three last year, four this year — traveling in a camper across Arizona, Utah, Colorado, and California. Second guessing myself during these trips has become the norm for me. But I try to make our camping trips as memorable and safe as possible.

How I’m encouraging exercise for my child with cystic fibrosis
Prepare for the expected care routine.
When we travel , I start preparing weeks before by checking our stock of medications . This year, I had to request a vacation override from our insurance to cover a two-month supply. That process alone took several days to wade through.
Next up is list making. Beyond the medications, we also need other supplies to care for Claire, our daughter with CF, and they’re not readily available in many destinations. Currently, most of her prescriptions are filled through a delivery speciality pharmacy, which I don’t have access to while traveling. For that reason, I have to check and recheck that we have ample supplies for all of our daughter’s care on the road.
With camping, however, I also have to keep supplies to a minimum. Space is limited. I have to stay organized in a small space with four people. It’s important that I make sure I check what’s absolutely necessary and leave the ancillary items. I sometimes miss the comforts of breathing treatments at home. But while camping, we have to stick to the bare necessities.
Do your research
Our family loves to do special activities while camping, including fishing, horseback riding, and hiking. Most of these involve some level of risk for a person with cystic fibrosis, given the environmental factors. As a result, I have to do research to see if Claire can participate and how I can minimize risk. If she can’t participate, I have decide whether we forgo it as a family or separate so some can enjoy the activity. It can be a reality check to make these choices, especially since camping is supposed to be carefree.
Last year, my kids were begging me to go to a hot springs pool they saw last summer. I’d told them absolutely not as we drove by, picturing all the possible bacteria floating in its waters. I was completely unprepared and later felt bad about how rigid I was.
For this year’s trip, which is still underway, I knew we’d again be staying near the hot springs pool, so I called the operators ahead of our visit and asked questions about their cleaning process and chlorination system. After that research, I felt much better making an informed decision. As a parent, I also felt better giving my children a more conscious answer. Anticipating activities helps, but with camping, we always have to expect the unexpected.
When surprises happen
I learned that lesson this trip when Claire spiked a high fever while we were in a rural location. I knew a CF clinic was five hours away, but I hadn’t done any research on the closest hospitals or checked updated road conditions. At 2 a.m., I found myself searching for accessible emergency room options with a fever-ridden child. It was terrifying, but it taught me that no one can plan for every scenario.
Part of camping is being ready to adapt in the face of unexpected bumps in the road. Luckily, being a cystic fibrosis parent grants you a steep learning curve of risk assessment , researching, and resourcing quickly. While it’s scary camping with a child who has CF, Claire is my outdoor girl. Seeing her joy in nature helps me push through the anxieties I have.
Note: Cystic Fibrosis News Today is strictly a news and information website about the disease. It does not provide medical advice, diagnosis , or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website. The opinions expressed in this column are not those of Cystic Fibrosis News Today or its parent company, Bionews, and are intended to spark discussion about issues pertaining to cystic fibrosis.
About the Author

Leave a comment
Fill in the required fields to post. Your email address will not be published.
Your CF Community

Recent Posts
- Lung transplants for CF on decline since Trikafta: U.S. study July 19, 2024
- Type 2 inflammation in CF linked to worse lung function July 18, 2024
- Sionna expands its pipeline of CFTR modulators with AbbVie’s assets July 17, 2024
- Adventures in camping outdoors with a child who has cystic fibrosis July 16, 2024
- Chronic rhinosinusitis in mom and daughter marked by CF symptoms July 16, 2024
Recommended reading

Type 2 inflammation in CF linked to worse lung function

Sionna expands its pipeline of CFTR modulators with AbbVie’s assets

Chronic rhinosinusitis in mom and daughter marked by CF symptoms
Subscribe to our newsletter.
Get regular updates to your inbox.
- Fierce Pharma
- Fierce Biotech
- Fierce Healthcare
- Fierce Life Sciences Events
- Cell & Gene Therapy
- Clinical Data
- Venture Capital
- Diagnostics
- AI and Machine Learning
- Special Reports
- Special Report
- Awards Gala
- Fierce Events
- Industry Events
- Whitepapers
Sionna salvages AbbVie assets, teeing up dual combo assault on cystic fibrosis
Sionna Therapeutics has quickly found a use for some of its $182 million series C haul, inking a deal with AbbVie to add three clinical-phase candidates to its burgeoning pipeline of cystic fibrosis prospects.
Boston-based Sionna has established itself as a potential threat to Vertex’s iron-grip on the cystic fibrosis market by advancing programs directly targeting correction of NBD1. The biotech has identified NBD1 correction as a way to fully restore CFTR function in patients with the most common mutation of the key cystic fibrosis protein. But it has also recognized combinations may be the route to the best results.
Sionna fast tracked its combination ambitions Tuesday by picking up a trio of molecules from AbbVie. The acquired portfolio includes two midphase compounds, the CFTR corrector ABBV-2222 and the CFTR potentiator ABBV-3067.
AbbVie studied ABBV-2222 and ABBV-3067, molecules respectively also called galicaftor and navocaftor, in combination with a C1 or C2 inhibitor in a midphase clinical trial. However, neither combination lived up to AbbVie’s expectations, prompting the company to stop the study and ax its cystic fibrosis program. AbbVie expanded in cystic fibrosis by paying Galapagos $45 million upfront in 2018.
Sionna has also picked up the rights to a phase 1 corrector, ABBV-2851. The biotech said ABBV-2222 and ABBV-2851 are correctors directed at TMD1, one of the CTFR protein function domains. Sionna has its own TMD1-directed corrector, SION-676, in preclinical development, plus an ICL4-targeted candidate, SION-109, in phase 1.
The expansion of the pipeline gives Sionna a choice to make. Pairing Sionna’s NBD1 stabilizers with any one of SION-109 and the three ex-AbbVie assets delivered better efficacy than the standard of care in a cystic fibrosis assay, the biotech said. Sionna will prioritize advancing one of the four compounds in a dual combination with its most advanced NBD1 stabilizer.
AbbVie parted company with the assets in return for an upfront payment, an equity investment in Sionna and a chance to receive late-stage development and commercial milestones and royalties. Neither party has disclosed the size of the deal.
An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- v.15(9); 2023 Sep
- PMC10575793
Uncommon Presentation of Cystic Fibrosis: A Case Report and Literature Review
Majed abu sirhan.
1 Pediatric Department, Barzilai University Medical Center, Ashqelon, ISR
Michael Kalinin
2 Pediatric Intensive Care Unit, Barzilai University Medical Center, Ashqelon, ISR
3 Genetics Institute, Barzilai University Medical Center, Ashqelon, ISR
4 Faculty of Health Sciences, Ben-Gurion University of the Negev, Beersheva, ISR
Evgenia Gurevich
Cystic fibrosis (CF) is a multiorgan disease, caused by autosomal recessive (AR) mutations in the cystic fibrosis transmembrane regulator (CFTR) acting primarily as a chloride channel. CF is most commonly diagnosed in Caucasian populations. Common clinical presentations in pediatric patients include chronic cough, respiratory tract infections such as pneumonia, digestive symptoms, and stunted growth, and malnutrition due to gastrointestinal malabsorption and pancreatic insufficiency. Excessive sweat sodium chloride losses due to dysfunctional sweat glands in CFTR result in volume contraction and secondary hyperaldosteronism leading to renal potassium losses and metabolic alkalosis. Hypokalemic hypochloremic metabolic alkalosis is a known but uncommon presenting sign of the disease, documented as pseudo Bartter syndrome. Common mutations in the CFTR gene are now included in prenatal genetic screening programs. We describe the case of an infant of African descent with normal prenatal screening who presented with severe hypokalemic hypochloremic metabolic alkalosis and was diagnosed with CF with further genetic confirmation of the diagnosis.
Introduction
Cystic fibrosis (CF) is a multiorgan disease, caused by autosomal recessive (AR) mutations in the CFTR gene, which regulates the movement of chloride ions across cell membranes [ 1 ]. CF is most commonly diagnosed in Caucasian populations but can affect other ethnic groups as well, with an incidence of one in 4100 live births in the United States, one in 2500-3500 live births in individuals of European descent, and an average of one in 5000 in Israeli population depending on ethnic subgroups [ 2 , 3 ]. Common clinical presentations in pediatric patients include chronic cough, respiratory tract infections such as pneumonia, and digestive symptoms such as stunted growth and malnutrition due to gastrointestinal malabsorption and pancreatic insufficiency. Hypochloremic hypokalemic metabolic alkalosis is a known but uncommon presenting sign of the disease, documented as pseudo Barter syndrome [ 4 ]. It is caused by excessive sweat sodium chloride losses due to dysfunctional CFTR protein, leading to volume contraction, secondary hyperaldosteronism, and the development of hypokalemia and hypochloremic metabolic alkalosis. CF is now included in neonatal screening programs globally, allowing early detection, management, and improved clinical outcomes [ 5 ].
In this report, we describe a case of an infant of African descent with normal prenatal screening who presented with severe hypokalemic hypochloremic metabolic alkalosis and was diagnosed with CF with further genetic confirmation of the diagnosis.
Case presentation
A previously healthy five-month-old female presented in the pediatric emergency room with weight loss and dehydration without apparent cause, as there was no history of fever, vomiting, diarrhea, or poor feeding. She had been born in our hospital at term after normal pregnancy to healthy non-consanguineous parents of African descent. Prenatal carrier screening, which, in our country, includes common pathogenic variants in the CFTR gene (delF508, W1282X, N1303K, G542X,3849+10Kb, 1717+1G>A,3121-1G>A, Y1092X, I1234V, W1089X, S549R, G85E, 405+1G.A), was normal. She had no history of respiratory or gastrointestinal complaints, and her growth and development were normal. On admission, her physical examination was unremarkable except for moderate to severe dehydration signs such as dry mucous membranes, sunken eyes, decreased skin turgor, and decreased urine output. Her heart rate was 145/minute, blood pressure was normal (85/62 mmHg), her weight was 5.2 kg (third percentile), and after intensive rehydration with isotonic saline raised to 5.7 kg (10th percentile). Blood test results on admission revealed severe metabolic alkalosis (pH 7.68, partial pressure of carbon dioxide (PCO2) 30 mmHg, bicarbonate (HCO3) 35 mmol/L), hypochloremia (chloride (Cl) 73 mmol/L), severe hyponatremia (sodium (Na) 124 mmol/L), and hypokalemia (K 2.6 mmol/L). Using an algorithm for differential diagnosis of normotensive hypokalemic hypochloremic metabolic alkalosis, we ruled out urinary and stool chloride losses (urine chloride <15 mmol/L, stool chloride 12 mmol\L). A sweat chloride test was positive (125 mmol/L, Na<40 mmol/L), suggesting the diagnosis of CF. Laboratory data for our patient are presented in Table Table1 1 .
Na: sodium; K: potassium; Cl: chlorine; PCO2: partial pressure of carbon dioxide; HCO3: bicarbonate
| Result | Reference range | |
| Na blood (mmol/L) | 124 | 136-145 |
| K blood (mmol/L) | 2.6 | 3.5–5.10 |
| CI blood (mmol/L) | 73 | 98-107 |
| pH | 7.68 | 7.32–7.43 |
| PCO2 (mmHg) | 30 | 38-48 |
| HCO3 (mmol/L) | 35 | 22-29 |
| CI urine (mmol/L) | <15 | n/a |
| CI Stool (mmol/L) | 12 | n/a |
| Sweat CI (mmol/L) | 125 | <29 |
The results of the genetic panel for common CFTR mutations were negative. CFTR next-generation sequencing (NGS) revealed a homozygous missense variant in exon 4, specifically c.416A>T; p.His139Leu; chr7:117171095A>T, indicated as deleterious by several predictive bioinformatic tools.
The diagnostic algorithm for our patient is presented in Figure Figure1 1 .

During the hospital stay for five days, the patients' electrolyte abnormalities and dehydration were treated with isotonic saline and potassium supplementation. After normalization of electrolytes and blood gases and volume repletion, potassium supplementation was stopped, and the patient was discharged home with NaCl supplementation alone. Due to the patient's abnormal presentation, her four-year-old sister was brought to medical attention as she had a history of recurrent lung infections and failure to thrive. She underwent genetic testing for CF and the same mutation as in her sister was found. Both the patient and her sister were referred to the CF clinic in the tertiary medical center for further treatment and follow-up.
In this report, we describe the uncommon presentation of CF with hypokalemic hypochloremic metabolic alkalosis and the algorithm of the diagnosis in pediatric wards in an infant of African descent with normal prenatal screening results.
The approach to the differential diagnosis of metabolic alkalosis includes, as a first step, blood pressure measuring, which was normal in our patient. The differential diagnosis of normotensive hypokalemic hypochloremic metabolic alkalosis includes urinary chloride losses (Barter and Gitelman syndromes, diuretic use), gastrointestinal losses (vomiting, chloride diarrhea), and skin losses (CF). The use of this diagnostic algorithm in our case helped to make an appropriate diagnosis based on an abnormal sweat test after the exclusion of urinary and gastrointestinal chloride losses.
Metabolic alkalosis is an uncommon presentation of CF with only a few cases and case series previously described in the literature.
Beckerman et al. described a case series of 11 infants with CF, five of them were diagnosed between one and 12 months of age initially based on hypokalemia, hypochloremia, and metabolic alkalosis without major pulmonary and/or gastrointestinal symptoms [ 4 ]. In a case series of 103 children diagnosed with CF before the age of 12 months, 17 presented with hypokalemic hypochloremic metabolic alkalosis without prominent respiratory or gastrointestinal symptoms [ 6 ]. According to the study, predisposing factors for the development of metabolic alkalosis with hypoelectrolytemia in CF patients included early infant age, breastfeeding, delayed diagnosis, heat exhaustion, and the presence of CFTR mutations associated with a severe phenotype. Molecular screening for mutations in the CFTR gene in eight Sardinian children who presented with hypotonic dehydration and electrolyte imbalances, without pulmonary or pancreatic symptoms associated with CF, revealed the T3381 pathogenic variant in the CFTR gene, either in homozygosity or compound heterozygosity with another CF mutation. These findings highlight the unique relationship between this specific pathogenic variant and an uncommon presentation of CF with hypokalemic hypochloremic metabolic alkalosis [ 7 ]. In our patient, this unique presentation may be related to the extremely hot weather in the summertime in our country or possibly can be attributed to the specific mutation as described in the above case series.
CF is most commonly diagnosed in Caucasian populations. However, very little is known about CF in populations of African origin among whom it has been believed to be extremely rare [ 8 ]. Our patient was born to parents of African descent (Eritrean Ethiopian). Although consanguinity is uncommon in this population, which makes the possibility of the recessive homozygous disease extremely low, NGS analysis revealed a homozygous mutation in our patient. The identified variant has not been previously reported in the genome Aggregation Database (gnomAD) database (Broad Institute, Cambridge, Massachusetts). Several predictive bioinformatic tools have consistently indicated that this variant is likely to be deleterious. In ClinVar (National Center for Biotechnology Information, Bethesda, Maryland, United States), this variant has been reported three times ( {"type":"entrez-protein","attrs":{"text":"VCV53910","term_id":"1516833906","term_text":"VCV53910"}} VCV53910 ) [ 9 ]. This mutation is also documented in the CFTR Mutation Database of the Hospital for Sick Children [ 10 ], as well as in the Ethiopian population in The Israeli Medical Genetic Database [ 11 ]. Furthermore, this mutation has been predominantly detected in families originating from the Gulf area, such as native Saudis and Bahraini regions [ 12 ]. It appears to be a founder mutation in Western Asia and Eastern Africa. According to the Centre for Arab Genomic Studies, it ranks among the 10 most common CFTR variants in the Gulf area, with a frequency of 6-7% [ 1 , 12 ]. Moreover, it seems to be a founder mutation in Africa as well. Individuals carrying this mutation have exhibited significant pulmonary disease in infancy, failure to thrive, and pancreatic insufficiency. Consequently, in the case of the patient in the current study, validation through Sanger sequencing has been requested for both the affected individual and her sister.
Prenatal screening program now includes common mutations in the CFTR gene, but they may miss rare mutations [ 13 ]. Therefore, a normal genetic screening result does not definitively rule out the possibility of CF. Nonetheless, prenatal screening remains a valuable tool for early and accurate diagnosis. This case highlights the fact that prenatal screening is not diagnostic and underscores the importance of maintaining a high level of clinical suspicion for CF in infants who exhibit suspicious symptoms, regardless of their prenatal screening results or ethnicity.
Conclusions
This case report summarizes the diagnostic course in the pediatric ward to the final diagnosis of CF in an infant of African descent with uncommon presentation. Hypokalemic hypochloremic metabolic alkalosis may be the only presenting sign of CF. This diagnosis should be considered regardless of ethnicity and normal prenatal genetic testing.
The authors have declared that no competing interests exist.
Human Ethics
Consent was obtained or waived by all participants in this study
COMMENTS
Cystic fibrosis (CF) is a multisystem disorder caused by pathogenic mutations of the CFTR gene (CF transmembrane conductance regulator). Typical symptoms and signs include persistent pulmonary infection, pancreatic insufficiency, and elevated sweat chloride levels.
Although cystic fibrosis is progressive and requires daily care, people with CF are usually able to attend school and work. They often have a better quality of life than people with CF had in previous decades. Improvements in screening and treatments mean that people with CF now may live into their mid- to late 30s or 40s, and some are living into their 50s.
Since ancient times, children around the world have been afflicted with cystic fibrosis that leads to shortened lifespans. In medieval Europe, these children were believed to be cursed by witches and doomed to die. The curse that became folklore pronounced, "Woe to the child who tastes salty from a kiss on the brow, for he is cursed and soon will die." Salty skin was a sign of an impending ...
Treatment. There is no cure for cystic fibrosis, but treatment can ease symptoms, reduce complications and improve quality of life. Close monitoring and early, aggressive intervention is recommended to slow the progression of CF, which can lead to a longer life.. Managing cystic fibrosis is complex, so consider getting treatment at a center with a multispecialty team of doctors and medical ...
Cystic fibrosis (CF) is an inherited life-threatening disease that affects many organs. It causes changes in the electrolyte transport system. People with CF have problems in the glands that produce sweat and mucus. CF causes thick mucus that clogs certain organs such as the lungs, pancreas, and intestines. This may cause malnutrition, poor ...
Median age at diagnosis of cystic fibrosis is 6-8 months; two thirds of patients are diagnosed by 1 year of age. The age at diagnosis varies widely, however, as do the clinical presentation, severity of symptoms, and rate of disease progression in the organs involved.
How Cystic Fibrosis Is Diagnosed. Newborn screening. In the last decade, newborn screening has become standard and is now available in all 50 U.S. states. The newborn screen shows infants who have a high level of an enzyme called immunoreactive trypsin in their blood. This occurs when there is injury to the pancreas.
Cystic fibrosis is an autosomal recessive disease caused by mutations of the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR). ... Shen reported a 5.7‐year delay between the first clinical presentation and the eventual CF diagnosis, 45 while another study of pediatric Chinese patients revealed a median age at ...
Cystic Fibrosis - Etiology, pathophysiology, symptoms, signs, diagnosis & prognosis from the MSD Manuals - Medical Professional Version. ... May also be suggested by a positive prenatal screening test result, family history, or symptomatic presentation. Confirmed by a sweat test showing elevated sweat chloride on 2 occasions.
Cystic fibrosis (CF) is the most common lethal inherited disease in white persons. Cystic fibrosis is an autosomal recessive disorder, and most carriers of the gene are asymptomatic. News & Perspective Drugs & Diseases CME & Education Video Decision Point ... The clinical presentation, age at diagnosis, severity of symptoms, and rate of disease ...
Cystic fibrosis (CF) is one of the most commonly diagnosed genetic disorders. Clinical characteristics include progressive obstructive lung disease, sinusitis, exocrine pancreatic insufficiency leading to malabsorption and malnutrition, liver and pancreatic dysfunction, and male infertility. ... Clinical Presentations. The classic ...
Cystic fibrosis is a progressive, genetic disease that affects the lungs, pancreas, and other organs. There are close to 40,000 children and adults living with cystic fibrosis in the United States (and an estimated 105,000 people have been diagnosed with CF across 94 countries), and CF can affect people of every racial and ethnic group.
Cystic fibrosis (also known as CF or mucoviscidosis) is an autosomal recessive genetic disorder affecting most critically the lungs, and also the pancreas, liver, and intestine.
Introduction. Cystic fibrosis (CF) is an inherited disease affecting multiple organs. A genetic mutation results in thickened secretions which commonly leads to recurrent respiratory infections.. CF is the most common inherited disease in Caucasians. 1 In the UK there are around 10,600 cases, whereas worldwide there are thought to be around 100,000. 2
Cystic fibrosis tends to get worse over time and can be fatal if it leads to a serious infection or the lungs stop working properly. But people with cystic fibrosis are now living for longer because of advancements in treatment. Currently, about half of people with cystic fibrosis will live past the age of 40. Children born with the condition ...
Cystic fibrosis (CF) was first formally described in 1938 by the pathologist Dorothy Andersen, who recognized a number of patients with characteristic lesions of the pancreas and a clinical syndrome of failure to thrive, diarrhea, and recurrent respiratory infections. However, CF was recognized in European folklore hundreds of years earlier, and anticipated the sweat electrolyte abnormalities ...
Pregnant women may have a simple blood test to look for common mutations (abnormalities) in the CFTR gene that cause cystic fibrosis (CF). If she carries one CFTR gene mutation. There is a 50 percent chance that this CFTR gene mutation will be passed on to the baby. If the father is also a carrier of a CFTR gene mutation there is a 25 percent (1 in 4) chance that the baby will have CF.
View this table: Table 1 Presentation of cystic fibrosis (CF) Sweat chloride > 60 mmol/l is considered diagnostic of CF. Values of sweat chloride of 40-60 mmol/l are considered highly suggestive of CF, and in this situation the sweat test should be repeated and/or genetic testing conducted. Although in CF patients there is a fall in sweat ...
Background/aims: Cystic fibrosis is the most common inherited lethal disease, which could be frequently identified late in regions without newborn screening. There are dramatically better outcomes in the early diagnosis of cystic fibrosis patients. This study aimed to evaluate the spectrum of manifestations of cystic fibrosis at first admission leading to diagnosis.
Dillon has a classic presentation of cystic fibrosis (CF, "Appendix") . He also has the most common genetic cause of CF, two copies of F508del (a.k.a. Phe508del), a deletion of the amino acid phenylalanine at position 508 of the cystic fibrosis transmembrane conductance regulator (CFTR) protein. More than 2100 variations in the CFTR gene ...
Staphylococcus aureus is next to Pseudomonas aeruginosa the most isolated pathogen from the airways of cystic fibrosis (CF) patients, who are often infected by a dominant S. aureus clone for extended periods. To be able to persist, the pathogen has to adapt to the hostile niche of the airways to counteract host defence, antibiotic therapy and the competition with coinfecting pathogens.
Cystic fibrosis (CF) is the most common life-threatening autosomal recessive disease in the United States, occurring in approximately 1 in 3500 newborns. 1 - 3 Treatment advances over the past several decades have raised the median predicted survival age in the United States from the mid-teens in the 1970s to more than 36 years old today; 4 optimal outcomes depend on timely and accurate ...
1 INTRODUCTION. Cystic fibrosis (CF), a complex multisystem disorder, has witnessed remarkable advancements in treatment strategies over the past few decades, significantly enhancing patient outcomes and life expectancy. 1 Central to these advancements is the use of cystic fibrosis transmembrane conductance regulator (CFTR) modulators, which target the underlying genetic mutations responsible ...
Context: Lung transplantation extends survival for some patients with advanced cystic fibrosis, but it is complicated, has many potential risks, and its outcomes are difficult to predict. No standards exist for informed decision making about transplantation. Objective: To assess decision making from the perspective of caregivers of patients who faced the transplant decision before dying of ...
Cystic fibrosis is a hereditary disease that so far has been incurable. Those affected have thick, viscous mucus secretions in their lungs, and lung function diminishes steadily over time. Today ...
The opinions expressed in this column are not those of Cystic Fibrosis News Today or its parent company, Bionews, and are intended to spark discussion about issues pertaining to cystic fibrosis. Print This Page. About the Author. Jennifer Chamberlain Jennifer Chamberlain is a wife and the mother of two. Her youngest child was diagnosed with ...
State law requires newborn screening for 41 different conditions, including cystic fibrosis and certain other diseases, on all babies born in Ohio. The analysis takes place in ODH's Public ...
AbbVie expanded in cystic fibrosis by paying Galapagos $45 million upfront in 2018. Related. Sionna secures $182M series C to more than double its clinical-stage cystic fibrosis contenders.
Introduction. Cystic fibrosis (CF) is a multiorgan disease, caused by autosomal recessive (AR) mutations in the CFTR gene, which regulates the movement of chloride ions across cell membranes [].CF is most commonly diagnosed in Caucasian populations but can affect other ethnic groups as well, with an incidence of one in 4100 live births in the United States, one in 2500-3500 live births in ...
• Cystic Fibrosis (CF) molecular genetic results performed as part of the newborn screening pathway will no longer be visible in the patient's EPIC chart. A copy of these results will no longer be faxed to the Cystic Fibrosis Clinics with the preliminary CF newborn screen result. Confirmation testing on a diagnostic sample