
Case Studies
Meet the editorial board.

Irina Bancos
Rochester, MN, USA

Susan M. Webb
Barcelona, Spain

Iacopo Chiodini
Milan, Italy

An interactive case study on adrenal incidentalomas and mild autonomous cortisol secretion (MACS)
This case study ꟷ the Cushing’s Hub Clinical Case Competition 2023 winning entry ꟷ looks at the clinical evaluation and management of MACS ranging from the assessment of adrenal incidentalomas to the development of an individual treatment plan.
An interactive case study on ectopic Cushing’s syndrome
This interactive case study describes a case of ectopic Cushing’s syndrome in a middle-aged woman presenting with severe back pain.

An interactive case study on hidden hypercortisolism
This interactive case study describes an unusual case of Cushing’s syndrome in a largely asymptomatic, female patient presenting with a fragility fracture.

An interactive case study on Cushing´s syndrome due to primary bilateral macronodular adrenal hyperplasia
This case study—the Cushing’s Hub Clinical Case Competition winning entry—looks at primary bilateral macronodular adrenal hyperplasia as a cause of Cushing’s with special emphasis on the role of aberrant receptors.
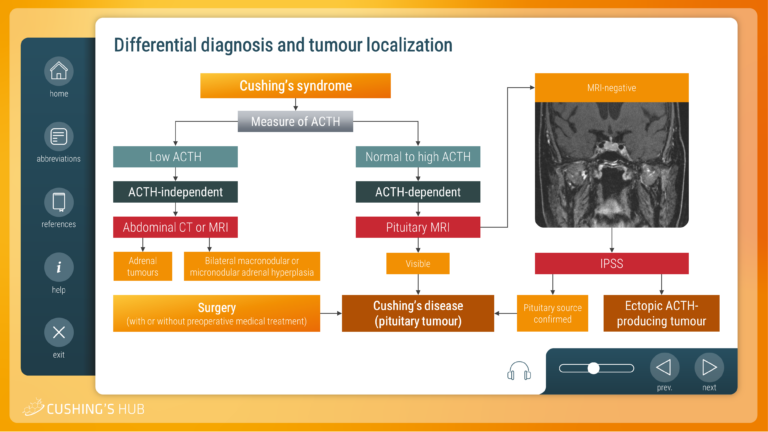
An interactive case study on the differential diagnosis of Cushing’s syndrome
This module looks at the diagnostic strategy required to establish the cause of adrenocorticotrophic hormone (ACTH)-dependent Cushing’s syndrome, when imaging and biochemical results are inconclusive.
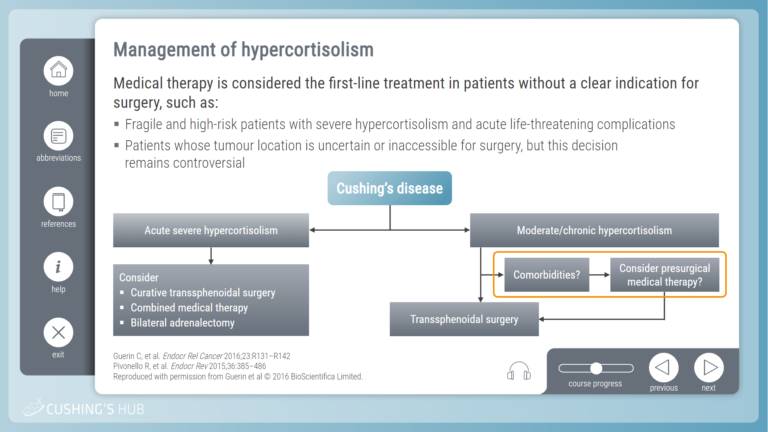
An interactive case study on severe hypokalaemia and excessive cortisol secreting Cushing’s disease
This module looks at the optimal way to manage hypercortisolism with associated comorbidities, particularly in patients who are ineligible for surgery.
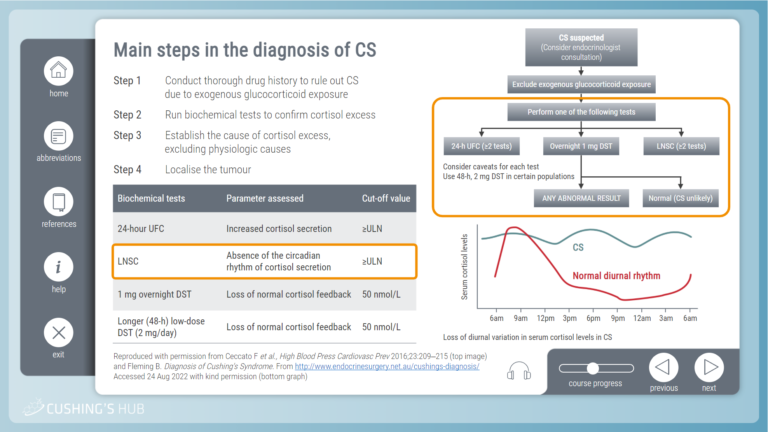
An interactive case study on screening for Cushing’s syndrome
This module raises questions about who should be screened for Cushing’s syndrome and considers the most suitable diagnostic tests for the avoidance of diagnostic uncertainty.
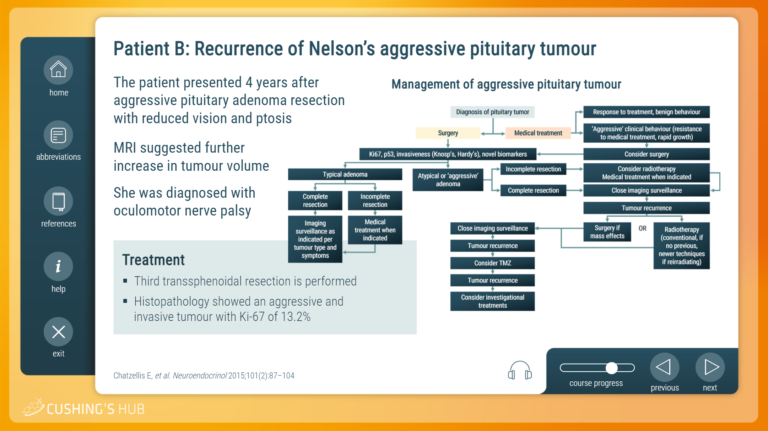
An interactive case study on the management of an aggressive Nelson’s tumour
The third interactive case study describes patient follow-up after bilateral adrenalectomy and explains how to develop a treatment plan for an aggressive Nelson’s tumour.
An interactive case study of relapsing Cushing’s disease in a woman of childbearing age
The second interactive case study explores the management of relapsing Cushing’s disease in a woman of childbearing age with emphasis given to pregnancy planning.

An interactive case study on cortisolemia
This interactive training module uses a case study format to explore the optimal way to assess cortisolemia in patients who have started treatment for Cushing’s syndrome.

© Springer Healthcare, 2024. All rights reserved.
This website is provided by springer healthcare, funded for educational purposes by hra pharma rare diseases., you are leaving cushings hub.
You are now leaving Cushings Hub and entering a website that we do not control.
Cushings Hub has provided this link for your convenience, but is not responsible for the content, links, privacy policy, or security of this website.
Click agree to proceed or close this window to return to Cushings Hub,
Before you go, why not bookmark Cushings Hub?
- Case report
- Open access
- Published: 23 February 2021
A challenging case of Cushing’s disease complicated with multiple thrombotic phenomena following trans-sphenoidal surgery; a case report
- Piyumi Sachindra Alwis Wijewickrama ORCID: orcid.org/0000-0001-7260-1727 1 ,
- Vithiya Ratnasamy 1 ,
- Noel P. Somasundaram 2 ,
- Manilka Sumanatilleke 2 &
- Sathyajith Buddhika Ambawatte 1
BMC Endocrine Disorders volume 21 , Article number: 29 ( 2021 ) Cite this article
3119 Accesses
3 Citations
Metrics details
Cushing’s syndrome occurs due to overproduction of cortisol from adrenal glands. Endogenous hypercortisolemia can occur secondary to adrenocorticotropic hormone (ACTH) dependent as well as independent causes. The presence of non-specific symptoms and signs contributes to a delay in diagnosis. Early identification and prompt definitive management is crucial. It is important to be alert about the post-operative complications including multiple thrombotic phenomena, which can add to the mortality. We report a case of Cushing’s disease in a young female managed with trans-sphenoidal surgery, followed by a challenging post-operative period complicated with multiple thrombotic phenomena, ultimately succumbed.
Case presentation
A 32-year-old Sri Lankan female presented with overt features of Cushing’s syndrome and diagnosed to have ACTH dependent Cushing’s disease with pituitary microadenoma. She underwent trans-sphenoidal surgery, following which she developed fatal multiple complications including diverticular rupture and ischemic colitis, needing hemicolectomy, followed by a parieto-occipital infarction.
This case highlights important and aggressive complications associated with Cushing’s syndrome giving rise to a challenging post-operative course. Diverticular rupture had been described in association with hypercortisolemia and this case adds to the existing literature. Post-operative ischemic colitis and stroke which contributed to the death of this patient could have been due to the procoagulant state associated with Cushing’s syndrome, with a high risk during the immediate post-operative period. This emphasizes the need to consider post-operative thromboprophylaxis in patients undergoing surgery for Cushing’s syndrome.
Peer Review reports
Cushing’s syndrome (CS) is caused by over production of cortisol from adrenal glands. This carries a very high morbidity and mortality due to associated complications if prompt diagnosis and effective management is not carried out.
Endogenous hypercortisolism is categorized as adrenocorticotropic hormone (ACTH) dependent and independent causes. ACTH dependent CS contributes to 80–85%, out of which Cushing’s disease (CD) due to pituitary ACTH hypersecretion is the most common, contributing to 75–80%, while 15–20% are due to ectopic ACTH syndrome [ 1 ]. ACTH independent CS due to adrenal adenomas or adrenal carcinomas contribute to 15–20% of the cases [ 1 ]. Usually, pituitary adenomas are microadenomas with only 5–10% of macroadenomas [ 2 ].
Patients present with a variety of clinical manifestations most of which are common with other diseases as well, such as obesity, hypertension, impaired glucose tolerance and menstrual irregularities, making the clinical diagnosis challenging. However, the presence of more discriminatory features, including purple striae, plethora, proximal myopathy, easy bruising, thin skin and unexplained osteoporosis should prompt the clinician to evaluate further with initial investigations including urinary free cortisol, late night salivary cortisol, 1 mg overnight dexamethasone suppression test (ODST), and standard two-days 2 mg dexamethasone suppression test (LDDST) [ 1 , 3 ].
Once the diagnosis is achieved, further investigations should be carried out to establish the cause. Plasma ACTH can distinguish between ACTH dependent and ACTH independent causes. Magnetic resonance imaging (MRI) of pituitary, followed by inferior petrosal sinus sampling (IPSS) if necessary is warranted for further evaluation of ACTH dependent CS [ 1 , 4 ].
It is of utmost importance to promptly control the disease and proceed with definitive management in order to minimize the associated morbidity and mortality. Cardiovascular diseases, cerebrovascular events and infections are the key contributors to mortality associated with CS [ 5 ]. The first line of management of CD is the excision of pituitary tumor, preferably via trans-sphenoidal approach, which is known to result in an initial remission rate of 60–80% [ 1 ]. However, effective management becomes extremely challenging due to multiple associated complications specially the thrombotic events which are more prominent in the immediate post-operative period, requiring close monitoring.
We report a case of CD in a young female due to ACTH secreting pituitary microadenoma managed with trans-sphenoidal surgery, with a challenging post-operative period due to multiple complications including thrombotic phenomena, ultimately succumbed.
A 32-year-old Sri Lankan teacher, mother of one, presented with progressive weight gain and lower limb swelling for 6 months. She also noted gradual change in her appearance with hyperpigmentation, acne and facial swelling. She had lower limb proximal muscle weakness and noticed recent memory impairment over the last 6 months which significantly affected her teaching activities. Her mentation was normal without suicide ideas or psychosis.
Her menstruation had been regular until 3 month ago, after which she became amenorrhoeic and pregnancy was excluded. She also experienced loss of libido. She gave a history of intermittent, vague generalized headache for 6 months duration, without associated visual disturbances. There was no galactorrhea. There was no back pain or fractures.
She was diagnosed with primary hypothyroidism 10 years ago and she was on levothyroxine 100 μg by the time she presented to us. She did not have a chronic cough, wheezing or flushing episodes. There was no history of exogenous steroid intake.
She has a 3-year-old child and there was no history of adverse pregnancy outcomes including miscarriages. There was no family history of similar illnesses.
On examination, she was obese with body mass index (BMI) of 38 kg/m 2 , with predominant central obesity and peripheral wasting. Her face was plethoric and round, with fat accumulation in cheeks and temporal areas, as well as dorsocervical and supraclavicular fat deposition giving rise to a thick, short neck. She had acne, hirsutism mainly involving face and upper body, as well as acanthosis nigricans. She had thin skin, ecchymoses at the venipuncture sites, together with wide, purplish striae over her upper arms and abdomen (Fig. 1 ). She had hyperpigmentation mainly involving the face and nailbeds. She had bilateral symmetric pitting lower limb edema up to mid-calf level without any tenderness or redness. There was no goiter. Her pulse rate was 88 beats per minute and blood pressure was 150/100 mmHg. Her respiratory and abdominal examinations were normal. She had reduced proximal muscle power in both upper and lower limbs. Her visual field examination was normal, as well as the fundi.

Examination findings supportive of Cushing's syndrome. a characteristic fat distribution in temporal region, cheeks and dorsal fat pad. b Facial plethora, hirsutism and acne. c Central obesity with purple striae over the abdomen
All her clinical manifestations were suggestive of CS. This patient had characteristic fat distribution supportive of CS, as well as discriminatory features including purple striae, proximal muscle weakness and ecchymoses which are more predictive, together with young hypertension.
Her initial investigations are summarized in Table 1 . Complete blood count revealed basophilia with low eosinophil count. Her potassium was low normal at 3.6 meq/L and she had high fasting blood glucose level, which further supported the diagnosis. Although the patient was initially treated for a primary hypothyroidism for 10 years, her current thyroid functions was suggestive of secondary hypothyroidism with inadequate thyroxine replacement, which is a common phenomenon seen in CS due to the suppression of thyroid stimulating hormone (TSH) pulse amplitude due to hypercortisolemia as well as due to blunted TSH response to thyroid releasing hormone [ 6 ].
Her ODST was positive at 728 nmol/L, suggestive of CS. ACTH was very high at 318 pg/mL, suggestive of ACTH dependent CS. High dose dexamethasone suppression test (HDDST) was 248 nmol/L showing a 50% suppression compared to baseline, indicating the likelihood of CD. Contrast enhanced computed tomography (CT) chest, abdomen and pelvis which was done to look for an ectopic source was negative apart from bilateral adrenal hyperplasia.
The MRI of pituitary revealed a high signal lesion of 10x8x9 mm in T1, T2 and FLAIR in the right side of pituitary, compatible with pituitary microadenoma (Fig. 2 ). IPSS revealed clear centralization and lateralization with a basal central to peripheral plasma ACTH ratio more than 2 and right side to left side inter sinus ratio of more than 1.4, confirming the source to be from right side of the pituitary (Table 2 ). Corticotrophin releasing hormone stimulation was not done due to unavailability of the reagent in local setting.

MRI showing high signal lesion of 10x8x9 mm suggestive of pituitary microadenoma. a T1 contrast sagittal. b T2 sagittal. c FLAIR axial
The patient was diagnosed with CD secondary to pituitary microadenoma. A trans-sphenoidal surgery was planned. Her disease was medically managed with ketoconazole 200 mg twice daily, with gradually increasing doses up to 400 mg twice daily, until the surgery, for 1 month. Controlling her CD burden was found to be extremely difficult, especially as ketoconazole was the only available medication at our setting which could be used for medical management of CS. Supportive management was done with anti-hypertensives, oral hypoglycemic drugs, and potassium supplements.
Within 1 month of diagnosis, she underwent endoscopic transnasal transsphenoidal hypophysectomy. Pituitary tumor was identified and removed by ring curettage and suction. Histology revealed pituitary adenoma with Ki67 proliferation index less than 1%.
Post- operatively, the patient was started on intravenous (IV) Hydrocortisone replacement of 50 mg 6 hourly, while continuing her thyroxine replacement. Serum 9 am cortisol on post-operative day 2 after withholding Hydrocortisone for 12 h came down to 80 nmol/L, indicating remission following surgery.
On post-operative day 5, patient complained of severe generalized abdominal pain. She had no associated vomiting and had opened bowel. On examination, her abdomen was distended, with generalized tenderness, without any palpable masses and intact bowel sounds. Patient had fever with temperature up to 102 degrees Fahrenheit. Lower limb examination did not reveal features suggestive of deep vein thrombosis. Her potassium was kept within the normal range with potassium supplements. She had neutrophilic leukocytosis with elevated C-reactive protein (CRP) at 56 mg/L. She was started on empirical IV antibiotics. However, she continued to have fever with CRP rising to 160 mg/L and continued to deteriorate with repeated X rays suggesting bowel perforation. Patient underwent an immediate explorative laparotomy, which revealed cecal perforation, after which right hemicolectomy with ileostomy and colostomy was done. Her clinical condition improved after the surgery.
Histology of right hemicolectomy specimen revealed cecal perforation with ruptured diverticulum, areas of hemorrhages, purulent material on serosal surface, thinned out, erythematous cecal wall with attenuated muscle wall and edematous submucosa. These morphological features were supportive of a ruptured diverticulum associated with serositis and ischemic colitis. The rest of the colonic mucosa was unremarkable without other diverticulae, crypt abscesses, dysplasia or malignancy.
Thereafter, on post-operative day 14 following the pituitary surgery, while the patient was being monitored in the intensive care unit, her conscious level suddenly deteriorated down to Glasgow Coma Scale (GCS) 7/15, and she was intubated and ventilated. The non-contrast CT (NCCT) brain revealed left parieto-occipital and cerebellar infarctions (Fig. 3 ). The patient continued to deteriorate, with further progression of infarcted area, and died 24 h later despite all attempts of resuscitation.

NCCT brain showing left sided parieto-occipital infarction, with involvement of the cerebellum
Discussion & conclusions
This case highlights the severity of complications associated with CD and that complications can even occur after surgical remission during immediate post-operative period following a prolonged disease course with poorly controlled hypercortisolemia.
The diagnosis of CD in this patient was straightforward and surgery was planned as the definitive therapy. Until the surgery, her disease was controlled with ketoconazole, a steroidogenesis inhibitor which is known to have a median response rate of 64% [ 7 ].
Following the trans-sphenoidal surgery her post-operative period became complicated despite achieving remission, with bowel perforation needing immediate laparotomy and hemicolectomy. Ischemic colitis with ruptured diverticulum were the most remarkable findings of the surgical specimens. Diverticular disease is widespread especially in the western countries. Usually, perforation of a diverticulum can occur due to high intracolonic pressure, disruption of colonic mucosal barrier, altered microflora and immunosuppression [ 8 ]. There is a well-known association between the treatment with exogenous corticosteroids and intestinal perforation. It was also previously observed that there is an association between intestinal perforation and endogenous glucocorticoid excess due to ACTH dependent CS [ 9 , 10 ]. Interestingly, most of these patients did not have a history of pre-existing diverticular disease. Several mechanisms are responsible for the diverticular perforation associated with hypercortisolism, including reduced collagen turnover leading to weakened colonic wall integrity, reduction of prostacyclin formation and impaired wound healing. Glucocorticoid-induced activation of tumour necrosis factor alpha receptors may play a role [ 9 ]. Cortisol excess is also known to cause a delay in diagnosis of bowel perforation by masking typical symptoms. In all the patients who were studied, it was evident that the intestinal perforation occurred when they were hypercortisolemic and not when they were in remission. In contrast, our patient developed this intestinal perforation with diverticular rupture following the pituitary surgery when biochemically proven to be in remission, suggesting a possibility of the impact from long-lasting excess cortisol prior to achieving remission. The prolonged disease course prior to surgery with severe hypercortisolemia could have contributed to this.
Features of ischemic colitis were also present in the hemicolectomy specimen. This can be attributed to the thrombotic tendency seen in patients with CS, which has manifested as mesenteric ischemia. Thromboembolic complications are four-fold higher among patients with CS [ 11 ]. This is due to increased synthesis of fibrinogen and von Willebrand factor stimulated by cortisol, as well as increased synthesis of plasminogen activator inhibitor type 1 [ 12 , 13 ]. Venous thrombo-embolism is the most commonly reported thrombotic phenomenon in these patients while acute mesenteric ischemia as seen in this patient is only rarely reported [ 14 ].
This patient also developed a left parieto-occipital stroke post-operatively, which may have largely contributed to her death. Cerebrovascular accident is a well-known complication in patients with CS, possibly contributed by the increased metabolic disease risk as well as procoagulant state. Patients with CS are known to be at high risk of stroke even before the diagnosis and the risk is known to remain elevated in long term follow up [ 15 ]. Vascular disease has been identified as the most common cause for death among patients with CS [ 5 ]. Furthermore, the involvement of multiple arterial territories suggests a possibility of underlying thromboembolic phenomena.
Clear guidelines do not exist regarding thromboprophylaxis in patients with CS. The Endocrine Society Clinical Practice guidelines suggest considering anticoagulation treatment peri-operatively specially as the risk of thrombo-embolism is highest in the first 4 weeks after surgery, due to worsening of the clotting profile [ 16 , 17 ]. The sudden reduction of cortisol level with its anti-inflammatory activity, leading to increased risk of inflammation and thrombotic state also contributes to the post-surgical worsening of the thrombotic risk [ 12 ]. This could have been a contributory factor for these complications in our patient, as she had normal post-operative cortisol level.
In a retrospective analysis of 313 patients with CS, it was found that, before the introduction of prophylactic anti-coagulation, 10% of patients with CS died due to thrombo-embolism and 10% had vascular morbidity, while the introduction of prophylactic anticoagulation reduced the morbidity due to thromboembolic events to 6% and mortality to 0.4% [ 13 ]. Therefore, it is rational to treat these patients with thromboprophylaxis in the immediate post-operative period where the thrombo-embolic risk will be highest due to reduced ambulation, as well as drastic drop of cortisol level leading to a pro-inflammatory and a pro-thrombotic state [ 18 ].
Routine thromboprophylaxis for patients with CS had not been practiced in our center up to now. While further studies are needed to assess its benefits and risks, patient selection and the exact duration, post-operative thrombo-prophylaxis should be considered in patients with CS following surgery, especially in the presence of additional risk factors.
Overall, the time taken for diagnosis and attaining remission after the onset of symptoms have been identified as key factors which increase mortality in patients with CS suggesting that duration of hypercortisolemia is linked to increased mortality [ 19 ].
In conclusion, patients with CS are prone to multiple complications, contributing to increased mortality, out of which thrombo-embolic phenomena, especially during immediate post-operative period are well known. This case highlights the rare occurrence of mesenteric ischemia, diverticular rupture and stroke, complicating the post-operative period in a patient with CD, ultimately leading to death. Therefore, this emphasizes the need of the prompt diagnosis and treatment of CS, as well as the need for prophylactic anticoagulation in patients undergoing surgery for CS.
Availability of data and materials
The data used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
Adrenocorticotropic hormone
Cushing's syndrome
Cushing's disease
Overnight dexamethasone suppression test
Low dose dexamethasone suppression test
Magnetic Resonance Imaging
Inferior petrosal sinus sampling
Body mass index
Fasting blood glucose
Thyroid stimulating hormone
Thyroid releasing hormone
High dose dexamethasone suppression test
Corticotrophin releasing hormone
Intravenous
C-reactive protein
Glasgow Coma Scale
Non-contrast CT
Sharma ST, Nieman LK, Feelders RA. Cushing’s syndrome: epidemiology and developments in disease management. Clin Epidemiol. 2015;7:281–93 Available from: https://www.ncbi.nlm.nih.gov/pubmed/25945066 .
PubMed PubMed Central Google Scholar
Tritos NA, Biller BMK, Swearingen B. Management of Cushing disease. Nat Rev Endocrinol. 2011;7:279. Available from. https://doi.org/10.1038/nrendo.2011.12 .
Article CAS PubMed Google Scholar
Nieman LK, Biller BMK, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The Diagnosis of Cushing’s Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93(5):1526–40. Available from. https://doi.org/10.1210/jc.2008-0125 .
Article CAS PubMed PubMed Central Google Scholar
Newell-Price J, Trainer P, Besser M, Grossman A. The Diagnosis and Differential Diagnosis of Cushing’s Syndrome and Pseudo-Cushing’s States. Endocr Rev. 1998;19(5):647–72. Available from. https://doi.org/10.1210/edrv.19.5.0346 .
Yaneva M, Kalinov K, Zacharieva S. Mortality in Cushing’s syndrome: data from 386 patients from a single tertiary referral center. Eur J Endocrinol. 2013;169(5):621–7 Available from: https://eje.bioscientifica.com/view/journals/eje/169/5/621.xml .
Article CAS Google Scholar
Dogansen SC, Yalin GY, Canbaz B, Tanrikulu S, Yarman S. Dynamic changes of central thyroid functions in the management of Cushing’s syndrome. Arch Endocrinol Metab. 2018;62:164–71.
PubMed Google Scholar
Pivonello R, De Martino MC, De Leo M, Simeoli C, Colao A. Cushing’s disease: the burden of illness. Endocrine. 2017;56(1):10–8. Available from. https://doi.org/10.1007/s12020-016-0984-8 .
Morris CR, Harvey IM, Stebbings WSL, Speakman CTM, Kennedy HJ, Hart AR. Epidemiology of perforated colonic diverticular disease. Postgrad Med J. 2002;78(925):654–8 Available from: http://pmj.bmj.com/content/78/925/654.abstract .
Shahidi M, Phillips RA, Chik CL. Intestinal Perforation in ACTH-Dependent Cushing’s Syndromeo Title. BioMed Res Int. 2019;2019:9721781.
Article Google Scholar
Sater ZA, Jha S, McGlotten R, Hartley I, El Lakis M, Araque KA, et al. Diverticular Perforation: A Fatal Complication to Forestall in Cushing Syndrome. J Clin Endocrinol Metab. 2018;103(8):2811–4. Available from. https://doi.org/10.1210/jc.2018-00829 .
Article PubMed PubMed Central Google Scholar
Jacoby RC, Owings JT, Ortega T, Gosselin R, Feldman EC. Biochemical Basis for the Hypercoagulable State Seen in Cushing Syndrome. Arch Surg. 2001;136(9):1003–7. Available from. https://doi.org/10.1001/archsurg.136.9.1003 .
Wagner J, Langlois F, Lim DST, McCartney S, Fleseriu M. Hypercoagulability and Risk of Venous Thromboembolic Events in Endogenous Cushing’s Syndrome: A Systematic Meta-Analysis. Front Endocrinol (Lausanne). 2019;9:805 Available from: https://www.ncbi.nlm.nih.gov/pubmed/30745894 .
Boscaro M, Sonino N, Scarda A, Barzon L, Fallo F, Sartori MT, et al. Anticoagulant prophylaxis markedly reduces thromboembolic complications in Cushing’s syndrome. J Clin Endocrinol Metab. 2002;87:3662–6.
CAS PubMed Google Scholar
Takayasu S, Murasawa S, Yamagata S, Kageyama K, Nigawara T, Watanuki Y, et al. Acute mesenteric ischemia and hepatic infarction after treatment of ectopic Cushing’s syndrome. Endocrinol Diabetes Metab Case Rep. 2017;2017:16–144 Available from: https://www.ncbi.nlm.nih.gov/pubmed/28480039 .
Dekkers OM, Horváth-Puhó E, Jørgensen JOL, Cannegieter SC, Ehrenstein V, Vandenbroucke JP, et al. Multisystem Morbidity and Mortality in Cushing’s Syndrome: A Cohort Study. J Clin Endocrinol Metab. 2013;98(6):2277–84. Available from. https://doi.org/10.1210/jc.2012-3582 .
Nieman LK, Biller BMK, Findling JW, Murad MH, Newell-Price J, Savage MO, et al. Treatment of Cushing’s Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2015;100(8):2807–31. Available from. https://doi.org/10.1210/jc.2015-1818 .
Casonato A, Pontara E, Boscaro M, Sonino N, Sartorello F, Ferasin S, et al. Abnormalities of von Willebrand factor are also part of the prothrombotic state of Cushing’s syndrome. Blood Coagul Fibrinolysis. 1999;10(3):145–51 Available from: http://europepmc.org/abstract/MED/10357009 .
Stuijver DJ, van Zaane B, Feelders RA, Debeij J, Cannegieter SC, Hermus AR, et al. Incidence of venous thromboembolism in patients with Cushing’s syndrome: a multicenter cohort study. J Clin Endocrinol Metab. 2011;96:3525–32.
Lindholm J, Juul S, Jørgensen JOL, Astrup J, Bjerre P, Feldt-Rasmussen U, et al. Incidence and Late Prognosis of Cushing’s Syndrome: A Population-Based Study1. J Clin Endocrinol Metab. 2001;86(1):117–23. Available from. https://doi.org/10.1210/jcem.86.1.7093 .
Download references
Acknowledgements
Author information, authors and affiliations.
Ministry of Health, Colombo, Sri Lanka
Piyumi Sachindra Alwis Wijewickrama, Vithiya Ratnasamy & Sathyajith Buddhika Ambawatte
National Hospital of Sri Lanka, Colombo, Sri Lanka
Noel P. Somasundaram & Manilka Sumanatilleke
You can also search for this author in PubMed Google Scholar
Contributions
PW – Involved in diagnosis and management of the patient and played a major role in writing the final manuscript. VR – involved in management of the patient and writing the final manuscript. NS – involved in diagnosis and management of the patient and in writing the final manuscript. MS - involved in diagnosis and management of the patient and in writing the final manuscript. SA – Involved in writing the final manuscript. All authors have read and approved the manuscript.
Corresponding author
Correspondence to Piyumi Sachindra Alwis Wijewickrama .
Ethics declarations
Ethics approval, consent for publication.
Written consent for publication and use of the images were obtained from the patient’s husband as the patient had passed away.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
Wijewickrama, P.S.A., Ratnasamy, V., Somasundaram, N.P. et al. A challenging case of Cushing’s disease complicated with multiple thrombotic phenomena following trans-sphenoidal surgery; a case report. BMC Endocr Disord 21 , 29 (2021). https://doi.org/10.1186/s12902-021-00701-0
Download citation
Received : 29 December 2020
Accepted : 16 February 2021
Published : 23 February 2021
DOI : https://doi.org/10.1186/s12902-021-00701-0
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Cushing’s disease
- Diverticular rupture
BMC Endocrine Disorders
ISSN: 1472-6823
- General enquiries: [email protected]

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
- My Bibliography
- Collections
- Citation manager
Save citation to file
Email citation, add to collections.
- Create a new collection
- Add to an existing collection
Add to My Bibliography
Your saved search, create a file for external citation management software, your rss feed.
- Search in PubMed
- Search in NLM Catalog
- Add to Search
Early recognition of Cushing's disease: a case study
Affiliation.
- 1 (Nurse Practitioner), Department of Neurosurgery, Pituitary/Neuroendocrine Center, Brigham and Women's Hospital, Boston, Massachusetts (Professor), Department of Neurosurgery, Harvard Medical School, Boston, Massachusetts.
- PMID: 24170635
- DOI: 10.1111/1745-7599.12014
Purpose: To present a case study of a 34-year-old woman with Cushing's disease and provide nurse practitioners (NPs) with the understanding of the clinical presentation needed for early recognition and treatment of the disease.
Data sources: A comprehensive review of published literature on Cushing's disease. Findings from history, physical examination, and diagnostic studies of a woman presenting to primary care NPs, physicians and other healthcare providers with multiple symptoms of Cushing's disease.
Conclusions: Cushing's disease is the result of the pituitary gland producing excess amounts of adrenocorticotropic hormone (ACTH) causing the overproduction of cortisol. The disease is fairly rare and is seen mostly in women. Common chief complaints include increased facial hair, weight gain, amenorrhea, changes in the face, neck, and abdomen, with muscle wasting of the lower extremities. Untreated, diabetes mellitus and hypertension can occur and increase the patient's morbidity and mortality. Early recognition and appropriate referral can reverse the signs and symptoms over time and lead to a significantly improved quality of life.
Implications for practice: This case presented the challenges faced by NPs and physicians in diagnosing patients with Cushing's disease.
Keywords: Diabetes; endocrine; hormone; hypertension; obesity; pituitary; women.
©2013 The Author(s) ©2013 American Association of Nurse Practitioners.
PubMed Disclaimer
Similar articles
- Managing Cushing's disease: the state of the art. Colao A, Boscaro M, Ferone D, Casanueva FF. Colao A, et al. Endocrine. 2014 Sep;47(1):9-20. doi: 10.1007/s12020-013-0129-2. Epub 2014 Jan 11. Endocrine. 2014. PMID: 24415169 Review.
- Cushing's disease: a multidisciplinary overview of the clinical features, diagnosis, and treatment. Buliman A, Tataranu LG, Paun DL, Mirica A, Dumitrache C. Buliman A, et al. J Med Life. 2016 Jan-Mar;9(1):12-18. J Med Life. 2016. PMID: 27974908 Free PMC article. Review.
- Cushing’s disease and melancholia. Condren RM, Thakore JH. Condren RM, et al. Stress. 2001 Jun;4(2):91-119. doi: 10.3109/10253890109115725. Stress. 2001. PMID: 22432130 Review.
- Cushing's disease: pathobiology, diagnosis, and management. Lonser RR, Nieman L, Oldfield EH. Lonser RR, et al. J Neurosurg. 2017 Feb;126(2):404-417. doi: 10.3171/2016.1.JNS152119. Epub 2016 Apr 22. J Neurosurg. 2017. PMID: 27104844 Review.
- Cushing's disease: current medical therapies and molecular insights guiding future therapies. Lau D, Rutledge C, Aghi MK. Lau D, et al. Neurosurg Focus. 2015 Feb;38(2):E11. doi: 10.3171/2014.10.FOCUS14700. Neurosurg Focus. 2015. PMID: 25639313 Review.
- Clayton, R. N., Raskauskiene, D., Reulen, R. C., & Jones, P. W. (2011). Mortality and morbidity in Cushing's disease over 50 years in Stoke-on-Trent, UK: Audit and meta-analysis of literature. Journal of Clinical Endocrinology & Metabolism, 96(3), 632–642.
- Findling, J. W., & Raff, H. (2006). Clinical review: Cushing's syndrome: Important issues in diagnosis and management. Journal of Clinical Endocrinology & Metabolism, 91(10), 3746–3753.
- Fode, N. C., Laws, E. R., & Northcutt, R. C. (1983). Pituitary tumors and hypertension: Implications for neurosurgical nurses. Journal of Neurosurgical Nursing, 15(1), 33–35.
- Laws, E. R., Reitmeyer, M., Thapar, K., & Vance, M. L. (2002). Cushing's disease resulting from pituitary corticotrophic microadenoma. Neurochirurgie, 48, 294–299.
- Lindholm, J., Juul, S., Jorgensen, J.L., Astrup, J., Bjerre, P., Feldt-Ramussen, U., Hagen, C., Jorgensen, J., Kosteljanetz, M., Kristensen, L., Laurberg, P., Schmidt, K., & Weeke, J. (2001). Incidence and late prognosis of Cushing's syndrome: A population-based study. Journal of Clinical Endocrinology & Metabolism, 86(1), 117–123.
Publication types
- Search in MeSH
Related information
Linkout - more resources, full text sources.
- Ovid Technologies, Inc.
- Wolters Kluwer
Other Literature Sources
- scite Smart Citations

- Citation Manager
NCBI Literature Resources
MeSH PMC Bookshelf Disclaimer
The PubMed wordmark and PubMed logo are registered trademarks of the U.S. Department of Health and Human Services (HHS). Unauthorized use of these marks is strictly prohibited.
Endocrine Abstracts
- Issues/Conferences
- Our Services
SFEBES2017 ePoster Presentations Neuroendocrinology and Pituitary (23 abstracts)
Cushing's disease - Case report
Katy chisenga.
University of Cambridge, Cambridge, UK.
Introduction: Cushing’s syndrome is caused by an extended exposure to increased levels of endogenous or exogenous glucocorticoids. It is a syndrome that can be extremely challenging to diagnose as many symptoms and signs are also indications of other disease processes.
Case: A 76 year old man presented to hospital with a six month history of immobility and falls. Proximal muscle weakness was also noted. The patient then underwent a period of rehabilitation.
The patient had a history of type 2 diabetes mellitus, hypertension, congestive cardiac failure, combined B12 and folate deficiency, longterm suprapubic catheter due to urinary retention, urinary tract infections and a myocardial infarction.
A first set of investigations revealed a 24 hr urinary free cortisol of 206 nmol/24 hr (0–146/24 hr), an overnight dexamethasone suppression test of 1588 nmol/L and a low dose dexamethasone suppression test of 1131 nmol/L (<50 nmol/L). The ACTH level was revealed to be 139 pmol/L and an MRI scan revealed a left-sided pituitary adenoma. As such, a diagnosis of ACTH-dependent Cushing’s disease was made.
The patient was initially managed with metyrapone with a view to transphenoidal surgery. However the patient developed shortness of breath and worsening peripheral oedema. A chest X ray and echocardiogram revealed left ventricular failure and reduced systolic function, respectively. For this reason it was decided to medically optimize his congestive cardiac failure and hypercortisolaemia as an inpatient with progression to neurosurgery if he were to stabilize.
Discussion: Cushing’s disease is a rarity that can be difficult to diagnose due to the significant number of varied pathologies indicated by its signs and symptoms. This is an interesting case of Cushing’s disease as the levels of cortisol measured in the patient were incredibly high.
Society for Endocrinology BES 2017
Harrogate, UK 06 Nov 2017 - 08 Nov 2017
Browse other volumes
Article tools
My recent searches, my recently viewed abstracts, chisenga katy.

Endocrine Abstracts ISSN 1470-3947 (print) | ISSN 1479-6848 (online) © Bioscientifica 2024 | Privacy policy | Cookie settings
BiosciAbstracts
Bioscientifica Abstracts is the gateway to a series of products that provide a permanent, citable record of abstracts for biomedical and life science conferences.
Cushing’s Syndrome Case Study (60 min)
Watch More! Unlock the full videos with a FREE trial
Included In This Lesson
Study tools.
Access More! View the full outline and transcript with a FREE trial
Ms. Smith Is a 33 year old female who presents to her primary care provider for General muscle weakness and low back pain. She reports that this pain has been going on for about 3 months and the weakness has been getting worse over the last 2 weeks and she has been more fatigued with basic physical exertion. She reports getting “steroid injections” in her back previously, but they “didn’t last long at all”.
What further history questions should be asked of Ms. Smith?
- What medical history does she have?
- What medications does she take on a regular basis?
- What has she tried in the past for her back pain? What has worked?
- Is the pain associated with any specific activity or time of day? What makes it worse or makes it better?
Ms. Smith has a history of Type II Diabetes and Asthma, and has been taking inhaled corticosteroids for the past 6 years. She also reports reports having irregular menstrual cycles for the past 2 years accompanied by unexplained weight gain in her abdomen. Her previous provider told her she might have Polycystic Ovarian Syndrome.
What initial nursing assessments should be performed?
- Heart and lung sounds
- Assess skin condition
- Assess strength x 4 extremities
- Abdominal assessment
Ms. Smith’s Vital Signs were as follows:
Temp 98.8°F
The nurse notes purple/pink stretch marks on arms, abdomen, and thighs. Ms. Smith has multiple cuts and bruises on her arms. When asked how she got them, she says “my skin is just so thin these days”. She is obese with noticeable fatty deposits in the midsection and upper back.
What diagnostic testing do you anticipate for Ms. Smith?
- Complete Metabolic Panel – test electrolytes and kidney/liver function
- Hormone levels – estrogen, progesterone, testosterone, cortisol
- Complete Blood Count to evaluate immune system
- ESR and/or CRP to assess for inflammation
Ms. Smith is sent home with a pain reliever for her back pain while the laboratory results are processed. An ultrasound of her kidneys and ovaries is ordered, pending scheduling an appointment for next week. Two days later, lab values result and show the following:
Cortisol 28 mg/dL (H)
Glucose 265 mg/dL
K 3.3 mEq/L
Na 148 mg/dL
Ca 7.8 mg/dL
Testosterone levels elevated
Which finding(s) is/are concerning and need to be reported to the provider? Why?
- Hypokalemia and hypernatremia can be detrimental to the cardiovascular and neurological system and need to be addressed quickly
- The elevated blood sugar and elevated cortisol levels combined with the clinical findings suggest possible Cushing’s Syndrome
What do you believe is going on physiologically with Ms. Smith?
- Ms. Smith likely has developed Cushing’s Syndrome due to chronic use of corticosteroids.
- This causes the Adrenal Glands to over-respond, secreting excess glucocorticoids (hence the hyperglycemia and fat distribution), excess mineralocorticoids (hence the electrolyte abnormalities), and excess androgens (hence the elevated testosterone levels).
- The hypocalcemia can also cause osteoporosis or soft, fragile bones
The provider notifies Ms. Smith that she needs to be seen again ASAP for further diagnostic testing to rule out any cardiac abnormalities. He tells her to stop taking her inhaled corticosteroid and prescribes a different rescue inhaler for her asthma. He also tells her she needs to begin taking some supplements, including calcium and potassium
Why does Ms. Smith need to have her heart checked out? What test would they do?
- The hypokalemia can cause electrical abnormalities or arrhythmias
- She needs an EKG
Why does Ms. Smith need calcium supplements? What caused her hypocalcemia? How might this contribute to her back pain?
- Cushing’s Syndrome causes hypocalcemia
- Hypocalcemia can cause calcium to be pulled from the bones to compensate – this creates an osteoporotic situation
- This may be why her back hurts – it is taking the weight of her body onto the soft, porous bones
Why does Ms. Smith have to stop taking her inhaler?
- The chronic use of the inhaled corticosteroids is the likely culprit – she should refer to her PCP or pulmonologist for other options to manage her asthma
View the FULL Outline
When you start a FREE trial you gain access to the full outline as well as:
- SIMCLEX (NCLEX Simulator)
- 6,500+ Practice NCLEX Questions
- 2,000+ HD Videos
- 300+ Nursing Cheatsheets
“Would suggest to all nursing students . . . Guaranteed to ease the stress!”
Nursing Case Studies

This nursing case study course is designed to help nursing students build critical thinking. Each case study was written by experienced nurses with first hand knowledge of the “real-world” disease process. To help you increase your nursing clinical judgement (critical thinking), each unfolding nursing case study includes answers laid out by Blooms Taxonomy to help you see that you are progressing to clinical analysis.We encourage you to read the case study and really through the “critical thinking checks” as this is where the real learning occurs. If you get tripped up by a specific question, no worries, just dig into an associated lesson on the topic and reinforce your understanding. In the end, that is what nursing case studies are all about – growing in your clinical judgement.
Nursing Case Studies Introduction
Cardiac nursing case studies.
- 6 Questions
- 7 Questions
- 5 Questions
- 4 Questions
GI/GU Nursing Case Studies
- 2 Questions
- 8 Questions
Obstetrics Nursing Case Studies
Respiratory nursing case studies.
- 10 Questions
Pediatrics Nursing Case Studies
- 3 Questions
- 12 Questions
Neuro Nursing Case Studies
Mental health nursing case studies.
- 9 Questions
Metabolic/Endocrine Nursing Case Studies
Other nursing case studies.
- Search Menu
- Sign in through your institution
- Volume 8, Issue 8, August 2024 (In Progress)
- Volume 8, Issue 7, July 2024
- Diabetes, Pancreatic and Gastrointestinal Hormones
- Growth, Growth Hormone, and Growth Factors
- Hormones and Cancer
- Lipids and Cardiovascular
- Obesity and Adipocyte Biology
- Parathyroid, Bone, and Mineral Metabolism
- Pituitary and Neuroendocrinology
- Reproductive Biology and Sex-Based Medicine
- Signaling Pathways
- Advance Articles
- Expert Endocrine Consult Articles
- ENDO Meeting Abstracts
- Bone Health Research
- Obesity Research
- Thematic Issues
- Clinical Practice Guidelines
- Endocrine Reviews
- Endocrinology
- Journal of the Endocrine Society
- The Journal of Clinical Endocrinology & Metabolism
- JCEM Case Reports
- Molecular Endocrinology (Archives)
- Endocrine Society Journals
- Author Guidelines
- Submission Site
- Open Access
- Why Publish with the Endocrine Society?
- Advertising & Corporate Services
- Reprints, ePrints, Supplements
- About Journal of the Endocrine Society
- Editorial Board
- Author Resources
- Reviewer Resources
- Rights & Permissions
- Member Access
- Terms and Conditions
- Journals on Oxford Academic
- Books on Oxford Academic

Article Contents
- < Previous
A Case of Iatrogenic Cushing’s Syndrome
- Article contents
- Figures & tables
- Supplementary Data
Andrea Del Toro Diez, Michelle Marie Mangual Garcia, Jose M Garcia-Mateo, Ernesto Jose Sola Sanchez, A Case of Iatrogenic Cushing’s Syndrome, Journal of the Endocrine Society , Volume 5, Issue Supplement_1, April-May 2021, Page A103, https://doi.org/10.1210/jendso/bvab048.206
- Permissions Icon Permissions
Cushing’s syndrome (CS) is considered a rare disease. The most common cause is the exogenous use of glucocorticoids (GCs), which are often given within a controlled medical setting, but their factitious use is rare. Factitious CS is more common in females, young patients, those with psychiatric disorders, and those with contacts within the medical field. The diagnosis of CS is challenging because some features are non-specific and commonly present in the general population, such as obesity, depression, diabetes, hypertension (HTN), and low bone mineral density (BMD). A high suspicion is warranted. We present the case of a 47-year-old man with HTN, obesity, dyslipidemia, obstructive sleep apnea, and low BMD who complained of increased appetite, significant weight gain, fatigue, sleepiness, muscle weakness, and occasional facial flushing. Medications include Hydrochlorothiazide, Furosemide, Losartan, Atorvastatin, and Teriparatide. Vital signs were normal and body mass index was 41.9 kg/m 2 . He had a round face, central obesity, and wide purple striae in his abdomen. Dual-energy X-ray absorptiometry scan showed low BMD at spine. Laboratories revealed a glycated hemoglobin of 6.1%, late-night salivary cortisol of <0.03 mcg/dL, 24-hour urine free cortisol of 22.5 mcg/24hr, morning cortisol of 0.01 ug/mL, ACTH 23.5pg/mL, and dehydroepiandrosterone sulfate (DHEA-S) 35 mcg/dL. Our patient persistently denied use of exogenous GCs, but a urine synthetic GC screen disclosed a positive result for dexamethasone; levels at 1.1 mcg/dL. After an exhaustive conversation, our patient confessed to using over-the-counter dexamethasone 4mg to treat occasional muscle aches. ACTH is usually suppressed in factitious CS, but this was not our patient’s case, giving the appearance of ACTH-dependent hypercortisolism. This can lead to unnecessary diagnostic and therapeutic approaches. An unsuppressed ACTH could be due to an unreliable ACTH immunoassay or intermittent, instead of continuous, ingestion of GCs. A suppressed DHEA-S level, as seen in our patient, may provide the clue to exogenous GC use as the cause of CS. Our case is also rare because our patient is male, older, and not related to the medical field. Hypercortisolism must be detected and treated early due to its high morbidity and mortality. Several features may be reversed with treatment. The possibility of hypothalamic-pituitary-adrenal (HPA) axis suppression due to prolonged use of GCs, resulting in adrenal insufficiency (AI) should be considered. The prevalence of GC-induced AI ranges from 14–63%, with the highest risk in those with Cushingoid features and those receiving a dose equivalent to prednisone 20mg daily for more than three weeks. Sudden withdrawal of GCs should be avoided to prevent adrenal crisis. A tapering regimen should be adopted with subsequent biochemical testing of the HPA axis once GCs have been reduced to a physiologic dose.
| Month: | Total Views: |
|---|---|
| May 2021 | 8 |
| June 2021 | 8 |
| July 2021 | 9 |
| August 2021 | 11 |
| September 2021 | 10 |
| October 2021 | 25 |
| November 2021 | 18 |
| December 2021 | 29 |
| January 2022 | 9 |
| February 2022 | 12 |
| March 2022 | 22 |
| April 2022 | 17 |
| May 2022 | 26 |
| June 2022 | 23 |
| July 2022 | 15 |
| August 2022 | 16 |
| September 2022 | 13 |
| October 2022 | 31 |
| November 2022 | 24 |
| December 2022 | 36 |
| January 2023 | 19 |
| February 2023 | 19 |
| March 2023 | 17 |
| April 2023 | 24 |
| May 2023 | 23 |
| June 2023 | 16 |
| July 2023 | 15 |
| August 2023 | 12 |
| September 2023 | 18 |
| October 2023 | 8 |
| November 2023 | 40 |
| December 2023 | 19 |
| January 2024 | 17 |
| February 2024 | 13 |
| March 2024 | 20 |
| April 2024 | 17 |
| May 2024 | 19 |
| June 2024 | 9 |
| July 2024 | 6 |
Email alerts
Citing articles via.
- About the Endocrine Society
- Advertising and Corporate Services
- Journals Career Network
Affiliations
- Online ISSN 2472-1972
- Copyright © 2024 Endocrine Society
- About Oxford Academic
- Publish journals with us
- University press partners
- What we publish
- New features
- Open access
- Institutional account management
- Rights and permissions
- Get help with access
- Accessibility
- Advertising
- Media enquiries
- Oxford University Press
- Oxford Languages
- University of Oxford
Oxford University Press is a department of the University of Oxford. It furthers the University's objective of excellence in research, scholarship, and education by publishing worldwide
- Copyright © 2024 Oxford University Press
- Cookie settings
- Cookie policy
- Privacy policy
- Legal notice
This Feature Is Available To Subscribers Only
Sign In or Create an Account
This PDF is available to Subscribers Only
For full access to this pdf, sign in to an existing account, or purchase an annual subscription.
Journals | Policy | Permission Journal of Endocrinology & Metabolism
INFORMATION
- For Authors
- For Reviewers
- For Editors
- For Readers
- Admin-login
| MOST READ |
(For the last 6-month published articles)

Original Article Review Case Report Short Communication Letter to the Editor Editorial
| Thanks to Reviewers 2023 |
|
|
| INDEXED IN |

|
| ||||
|
|
|
| ||
| QUICK LINKS | |
|
|
|
|
| |
|
| |
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
| ARTICLE STATISTICS |
| Submission to First Decision |
| 25 Days |
|
|
| 37% |
|
|
| 38 Days |
| Average article statistics from the last 12 months data |

| COVID-19 RESEARCH |
| The COVID-19 outbreak presents the unprecedented challenge for world public and medical practitioners and health care providers, the post COVID-19 condition (or long COVID) includes long term symptoms which may persist for months or years after SARS-CoV-2 infection. We will consider submissions related to all aspects of COVID-19 and Long COVID, and process the manuscripts in priority. |
- Online-First
| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website https://www.jofem.org |
- Case Report
Volume 12, Number 1, February 2022, pages 40-48
Cushing’s Disease of Unknown Etiology: A Clinical Case Report
a Endocrine Department, Santa Maria Hospital and Lisbon Medical School, Lisbon, Portugal b Internal Medicine Department, Jacobi Medical Center and Albert Einstein College of Medicine, Bronx, NY, USA c Neurosurgery Department, Santa Maria Hospital and Lisbon Medical School, Lisbon, Portugal d Pathology Department, Santa Maria Hospital and Lisbon Medical School, Lisbon, Portugal e Corresponding Author: Joao Martin Martins, Endocrine Department, Santa Maria Hospital and Lisbon Medical School, Lisbon, Portugal
Manuscript submitted October 21, 2021, accepted December 9, 2021, published online February 15, 2022 Short title: Cushing’s Disease of Unknown Etiology doi: https://doi.org/10.14740/jem784
- Introduction
Cushing’s disease (CD) remains a diagnostic and therapeutic challenge. Different subtypes may be recognized that will offer insight in this complex situation. We describe an atypical case that we assume is a common variation albeit with no previous formal description. A young female patient presented with minimal clinical features of CD, but increased adrenocorticotropic hormone (ACTH) and cortisol levels, with maintained circadian rhythm that was not suppressed either after the rapid dexamethasone or the prolonged low-dose dexamethasone tests, but suppressed with the prolonged high-dose dexamethasone test and presented a flat ACTH and cortisol response after the corticotropin-releasing hormone (CRH) test. A diffuse enlarged pituitary gland with thickened pituitary stalk was present and a mixed corticotroph adenoma was removed. CD persisted despite now normal pituitary morphology, except for pituitary stalk widening. Plasma levels of CRH were low and no abnormalities were found in the coding region or flanking introns of glucocorticoid receptor (GCR) gene ( NR3C1 ). Somatostatin receptors were not present in the octreoscan, and treatment with cabergoline or somatostatin analogs was ineffective. Morbidity and mortality are increased in CD even in patients successfully treated and in remission. Despite early success in over 80% of the patients, in the long term CD recurs in almost 50% of the patients. Defining subtypes of CD may help elucidate mechanisms of the disease. We propose a new variant that we assume is common. Furthermore adaptation to chronic hypercortisolism is present.
Keywords: Cushing’s disease variant; Dexamethasone test; Corticotroph adenoma; GCR gene; Subtypes
Hypercortisolism is a common condition in everyday clinical practice, but most cases are either obvious or not immediately relevant by themselves: hypercortisolism due to the use of supraphysiologic doses of corticosteroids in the treatment of inflammatory and immune-mediated disorders [ 1 ], hypercortisolism in relation to the ectopic secretion of adrenocorticotropic hormone (ACTH) in common malignancies like the small cell adenocarcinoma of the lung or more rarely pancreatic neuroendocrine tumors [ 2 , 3 ], and subclinical hypercortisolism in regard to incidentally found adrenal nodules, although in this case, clinical importance is still a matter of ongoing debate [ 4 ].
Hypercortisolism may also be particularly common in selected groups of patients like those with diabetes mellitus (< 10%), high blood pressure (1%), osteoporosis (11%) or in women with irregular menses and hirsutism (1%) [ 5 - 9 ].
Outside those specific settings hypercortisolism is much less common, with two to three new cases per million per year [ 10 ]. Diagnostic approach always includes discriminating between ACTH-dependent (around 70% of all cases) and ACTH-independent Cushing’s syndrome (CS) [ 11 , 12 ], even if there is still a significant delay in diagnosis [ 13 ] and an increased morbidity and mortality before and even after diagnosis and treatment [ 14 ].
Cushing’s disease (CD) with an estimated incidence of around 1.5 per million per year, female preponderance (3:1) and peaking at ages between 25 and 45 years accounts for most of the cases of ACTH-dependent CS [ 10 , 15 , 16 ]. CD is due in most cases to a monoclonal pituitary microadenoma, sometimes with oncogenic triggering events in the cyclin or ubiquitin pathways, variable biological features including variable expression of corticotropin-releasing hormone (CRH), glucocorticoid and dopaminergic receptors and variable proopiomelanocortin processing [ 17 - 20 ]. Rarely it occurs in patients with multiple endocrine neoplasia type 1 or 4, familial isolated pituitary adenoma, McCune-Albright syndrome or Carney complex [ 15 , 16 , 20 ].
CD may in some cases be difficult to identify in magnetic resonance imaging (MRI) studies, requiring invasive and technically demanding petrosal sinus catheterization, and can only be cured by surgical removal generally using the transphenoidal route, with a success rate of about 60-70% in the long term in the best series [ 15 , 16 , 21 ].
Despite a standardized clinical diagnostic and treatment approach [ 22 - 24 ], CD may be difficult to diagnose and to treat; and several uncommon variants are recognized like cyclic CD [ 25 ], intermediate lobe corticotroph adenomas [ 26 ], mixed corticotroph adenomas [ 27 ], silent corticotroph macroadenomas [ 28 ] and even the exceedingly rare corticotroph carcinoma [ 29 ]. Also apparent CD because of CRH hypersecretion, either a hypothalamic tumor or a peripheral neuroendocrine tumor [ 30 ], and the classic Nelson’ syndrome [ 31 ] after bilateral adrenalectomy must be considered. The relations of these variants to the biological features of the tumor are not completely understood.
We now report what seems to be a common variant of CD, with distinctive clinical, analytical and imagiological findings, recurring after pituitary surgery, which presents as a clinical challenge and may be informative of the pathogenesis of CD in general. We assume this variant is commonly recognized by other groups, although we could not find a formal description in the literature. Furthermore adaptation to chronic low-grade hypercortisolism is apparent.
Investigations
The patient is a Caucasian female, aged 29, single, working as a practicing nurse in a public central hospital in Lisbon and living in the Setubal district, less than 50 km south of Lisbon.
Since 1 year before, the patient had complained of headaches. In fact these were old complaints and with the general characteristics of migraine, with a periodicity of about once every month outside the menstrual period, with weight gain of about 8 kg since she was aged 20, associated with carbohydrate craving during periods of job stress, with no overweight and without a central distribution pattern of body fat, and with body striae that were in fact very mild at the thighs, thin, white and more easily explained by the weight gain. The patient emphatically insisted she was always very tired and that her body shape was changing. There was no diabetes mellitus, nor symptoms like increased thirst and frequent urination. Blood pressure was normal without any medication or cardiovascular symptomatology; and menstrual cycles were regular under contraceptive pill that was interrupted 2 months before, without any complains about hirsutism or acne. There were no previous attempted pregnancies. No visual defects were apparent and no other neurologic complaints were expressed.
There was a past episode suggestive of acute pyelonephritis. Hypermetropia was corrected some years ago by ocular surgery. No other diseases or complaints were recorded.
The patient did not smoke, drank only socially (ethanol consumption < 40 g/week), and was not taking any regular medications.
Her father, aged 57 was healthy except for high blood pressure under medical treatment; and her mother, aged 56, had previous diagnosis of breast carcinoma and non-Hodgkin lymphoma, both cured. One older brother aged 35 was healthy. There was no family record for endocrine diseases, breast diseases or hematologic diseases in direct relatives.
Physical examination ( Fig. 1 ) revealed an apparently healthy young female who was anxious, but otherwise with no behavioral alterations. Body temperature was normal (36 °C), conjunctives were not pale, and there was no peripheral edema. Height was 174 cm without shoes, weight was 69 kg (without shoes or coats), body mass index 22.8 kg/m 2 , umbilical perimeter 79 cm, waist perimeter 101 cm, thigh perimeter 62 cm (umbilical/waist ratio: 0.78); blood pressure 130/75 mm Hg in the right arm supine position, heart rate 80/min, and breath rate was 16 respirations/min. Head and neck examination were normal without alopecia, hirsutism, or acne lesions, no neurologic abnormalities regarding the cranial nerves on crude examination; and the thyroid was not enlarged and presented no palpable thyroid nodules. Chest examination revealed normally developed adult breasts with no nipple discharge and no cutaneous abnormalities, and breath and heart sound were supple, regular and with no abnormal findings. Abdominal examination revealed no tenderness, without any palpable masses and bowel sounds were normally present; there were no abdominal striae . Members examination did not revealed any changes with normal muscular tone, no evidence for venous abnormalities; and arterial pulses were present, regular and symmetrical; only minimal thin white striae could be found at the upper thighs. Crude neurologic examination was normal regarding muscle power, reflexes, motility, equilibrium or motor coordination.
| |
Since she was a health professional working in a public central hospital in Lisbon, with easy access to medical care, a head MRI study had already been obtained that revealed a large intra-sellar mass with 19 × 13 mm, extending up until the optic nerves with no lateral invasion of the cavernous sinuses. That mass representing the whole pituitary gland was hypointense in both T1- and T2-weighted sections with homogenous uptake of the contrast. There was a widening of the pituitary stalk and the neurohypophysis could not be defined clearly ( Fig. 2 ).
Also at another institution a complete analytical blood panel had been obtained with no hematologic or biochemical routine abnormalities. However at 9:00 am, ACTH was 40 pg/mL (reference values (RV): 7 - 63), cortisol 25 µg/dL (RV: 10 - 20) and 24-h urinary cortisol 596 µg (RV: < 285). Normal values of growth hormone (GH), insulin-like growth factor 1 (IGF1), free T4 (FT4), thyroid-stimulating hormone (TSH), prolactin, follicle-stimulating hormone (FSH) and luteinizing hormone (LH) were found. An overnight dexamethasone test (1.0 mg orally (PO) at 24 h) had been performed with a cortisol value at 8:00 am next morning of 29 µg/dL. As an outpatient at that institution, a prolonged low-dose dexamethasone test (0.5 mg, every 6 h for 48 h) was obtained with a cortisol value of 39 µg/dL at 8:00 am after the last dose. A prolonged high-dose dexamethasone test (2 mg every 6 h for 48 h) test revealed a cortisol value of 2.9 µg/dL, 8:00 am after the last dose. Although done at another institution in the same city of Lisbon, the same analytical methods were used in our institution and are later described.
Since there was a marked discrepancy between clinical and analytical data, and the later were obtained on a non-supervised outpatient basis, we decided to repeat that evaluation as an inpatient.
Baseline routine analytical evaluation obtained in morning after the overnight fast revealed no change with normal hematological values without leukocytosis or eosinopenia, normal renal function with no electrolyte or mineral abnormalities, normal serum glucose, lipids and proteins, and normal hepatic enzymes. Chest X-ray and electrocardiogram (EKG) were normal. Baseline endocrine evaluation was normal as follows: T3: 80 ng/dL (RV: 80 - 200), T4: 6.4 ng/dL (RV: 5 - 14), TSH: 2.6 µU/mL (RV: 0.3 - 4.2), FT4: 1.22 ng/dL (RV: 0.9 - 1.7), thyroid peroxidase antibody (TPOAb): < 10 U/mL (RV: < 10), thyroglobulin antibody (TgAb): < 10 U/mL (RV: < 10), prolactin: 15 ng/mL (RV: 5 - 23), GH: 7.6 ng/mL (RV: < 10), IGF1: 431 ng/mL (RV: 117 - 329); IGFBP3: 5.8 ng/mL (RV: 3.5 - 7.6), FSH: 9 U/L (RV: 2.5 - 10), LH: 6 U/L (RV: 2 - 12.5), E2: 42 pg/mL (RV: 11 - 200), progesterone: 0.8 ng/mL (RV: 3.5 - 26), TT: 22 ng/dL (RV: 0 - 81), androstenedione: 2.7 ng/mL (RV: 0.3 - 3.3), sex hormone binding globulin (SHBG): 32 nmol/L (RV: 18 - 114); parathyroid hormone (PTH): 33 pg/mL (RV: 7 - 65); 25-hydroxyvitamin D: 22 ng/mL (RV: 20 - 80), insulin: 11 mU/mL (RV: 3 - 29); C-peptide: 1.7 ng/mL (RV: 1.1 - 5.0). Markers of bone remodeling were also normal or slightly increased: osteocalcin 14 ng/mL (RV: 3.1 - 13.7); N-terminal fragments of type 1 collagen propeptide (P1NP): 49 pg/mL (RV: < 30); C-terminal fragments of type 1 collagen (CTX): 0.45 ng/mL (RV: < 0.3). Nonspecific markers for neuroendocrine tumors were normal: chromogranin A: 1.9 (RV: < 3) and neuron-specific enolase: 21 (RV: < 18). Specifically and regarding the pituitary-adrenal axis the following parameters were obtained: ACTH: 48 pg/mL (RV: 7 - 63), cortisol: 20 µg/dL (RV: 10 - 20), dehydroepiandrosterone sulphate (DHEAS): 370 µg/dL (RV: 35 - 430); 17OHP: 2.2 ng/mL (RV: 0.2 - 4.7); S compound: 2.5 ng/mL (RV: < 8); aldosterone: 111 pg/mL (RV: 10 - 180); renin: 5.3 µU/mL (RV: 2 - 20), and 24-h urinary cortisol: 1,059 µg (RV: < 285).
All analytical measurements were obtained at the Clinical Pathology Department of the hospital using commercially available automated and standardized methods. Specifically electrochemoluminescence immunoassay (ECLIA) methods (Roche, Cobas 8000, Basel) were used for hormone measurements. Intra- and interassay variation coefficients were always below 10%. Reference values for the adult population are established by the Clinical Pathology Department and periodically revised to sustain clinical decisions. The Clinical Pathology Department is certified by the international standard ISO 9001:2015 and regularly participates in official quality control programs.
The circadian rhythm was evaluated followed by the rapid overnight dexamethasone (1.0 mg PO at 24 h) test ( Table 1 ). Next, the prolonged low (dexamethasone 0.5 mg 6/6 h for 48 h) and high (dexamethasone 2 mg 6/6 h for 48 h) tests were obtained sequentially ( Table 2 ). Finally a CRH test was obtained (hCRH 100 µg, intravenous (IV), bolus at time 0) ( Table 3 ).
| | Circadian Rhythm and Rapid Overnight Dexamethasone (1.0 mg PO at 24 h) Test |
| | Prolonged Low (Dexamethasone 0.5 mg 6/6 h for 48 h) and High (Dexamethasone 2 mg 6/6 h for 48 h) Tests |
| | CRH Test (hCRH 100 µg, IV, Bolus at Time 0) |
Abnormalities with possible pathogenic significance in the glucocorticoid receptor (GCR) coding gene ( NR3C1 ) at 5q31 were not detected (exons and flanking introns) by QXT, Agilent Technologies and next generation sequencing MiSeq, Illumina with NextGENe, SoftGenetics (CGC Genetics, Porto, Portugal). CRH measurement at a private institution revealed low/suppressed CRH levels < 1 ng/mL (RV: 0 - 3.5) (enzymatic immunoassay (EIA), Cusabio ® , Dr. Joaquim Chaves, Laboratorio de Analises Clinicas, Lisbon, Portugal).
Six months later a pituitary adenoma was removed by sublabial incision and the transphenoidal route. Surgery was uneventful. Pathological examination ( Fig. 3 ) revealed adenohypophysis and neurohypophysis fragments as well as fragments with the pattern of solid adenoma, some with eosinophilic and some with clear cytoplasm; no mitoses were found and anisokaryosis was mild to moderate; and Ki67 was less than 3%. Some cells were stained for ACTH by immunocytochemistry. On the fifth postsurgical day (8:00 am) the following values were obtained: ACTH 37 pg/mL and cortisol 9 µg/dL. Two weeks after surgery the values were as follows: ACTH 48 pg/mL and cortisol 25 µg/dL.
Follow-up and outcomes
Six months later, under no medication, the patient maintained the same complaints as before, and menstrual cycles had not resumed after suspending the contraceptive pill. These complaints even if with poor objective translation had a profound impact on the quality of life of the patient. Analytical evaluation revealed ACTH: 44 pg/mL, cortisol: 17 µg/dL, 24-h urinary cortisol: 364 µg. Cortisol post dexamethasone (1.0 mg) the previous night: 15 µg/dL.
A new MRI did not reveal any pituitary abnormalities except for the persistent pituitary stalk widening ( Fig. 4 ).
A head scan with the radiolabeled somatostatin analogue ( 111 In-pentetreotide) was obtained with negative results; but despite that the somatostatin analogue (octreotide acetate, Sandostatin LAR ® ) 20 mg and later 30 mg, once a month, intramuscular (IM) was began and maintained for 8 months. Evaluation then found ACTH: 36 pg/mL; cortisol: 19 µg/dL; and 24-h urinary cortisol: 384 µg. Gallbladder stones developed although asymptomatic and less than 15 mm. Somatostatin was interrupted and cabergoline 0.5 mg, twice a week was began and maintained for 6 months: ACTH: 43 pg/mL; cortisol: 23 µg/dL; 24-h urinary cortisol: 158 µg; after 1 year ACTH: 44 pg/mL, cortisol: 17 µg/dL, 24-h urinary cortisol: 538 µg, and cortisol post dexamethasone (1.0 mg, PO) previous night: 27 µg/dL. Imipramine 25 mg, once daily at night was also attempted; and after 3 months it was revealed that ACTH: 72 pg/mL; cortisol: 16 µg/dL; 24-h urinary cortisol: 599 µg, and cortisol post dexamethasone (1.0 mg, PO): 14 µg/dL.
This is indeed a very atypical case of CD. Despite patient insistence, there was no objective clinical evidence for hypercortisolism except for amenorrhea that could only later be defined and maybe emotional changes that are common in patients with CD [ 12 , 16 , 32 , 33 ]. There was no central distribution of body fat or protein wasting namely proximal myopathy or purple wide skin striae that strongly characterizes hypercortisolism; there was no diabetes mellitus or glucose intolerance, no high blood pressure or dyslipidemia and no osteopenia or osteoporosis, no record for previous fractures or thrombotic events; there was no hirsutism or acne; there were no visual field defects, and headaches could be more easily explained by migraine [ 12 , 16 ].
Despite lack of objective clinical evidence for hypercortisolism and again emphasizing the truism that “the patient is always right”, analytical evidence repeatedly and unequivocally revealed hypercortisolism (see 24-h urinary cortisol). Interestingly enough the circadian rhythm was almost maintained since values at 19 - 20 h are about half of those at 8 - 9 h, a very rare finding in CD; midnight cortisol values were however unequivocally increased [ 12 , 16 , 22 , 23 ].
Furthermore and following a standardized diagnostic approach hypercortisolism was non suppressible by dexamethasone overnight; hypercortisolism was ACTH-dependent, and hypercortisolism was also not suppressed by the prolonged low-dose dexamethasone test, essentially excluding pseudo Cushing states like those of major depression and chronic stress [ 12 , 16 , 22 , 23 , 34 , 35 ], but suppressed by the high-dose prolonged dexamethasone test, essentially excluding ectopic ACTH secretion [ 22 , 23 , 36 ].
New diagnostic approaches to the differential diagnosis of CS emphasize the central role of the CRH or desmopressin test [ 37 ]. In the CRH test, a 30-40% increase in ACTH and a 20% increase in cortisol are generally used to define CD; while in ectopic CS baseline ACTH and cortisol values are generally much higher and the response is much less; and in chronic CRH oversecretion a decreased ACTH response and increased cortisol response are described. By these criteria, in this case the CRH test suggests CD albeit imperfectly, and is strongly against the other possibilities [ 12 , 16 , 22 , 23 , 37 - 40 ].
Common pitfalls in the diagnosis of CD do not seem to explain discrepancies; free cortisol is increased as evaluated by the 24-h urinary cortisol excluding cortisol-binding globulin (CBG) increase induced by pill; no drugs were used that could increase the metabolism of dexamethasone or present anti-glucocorticoid effects [ 12 , 16 , 22 , 23 , 37 ].
A certain degree of peripheral cortisol resistance is certainly suggested by the lack of clinical features of hypercortisolism despite analytical hypercortisolism, but familial syndromes of cortisol resistance do not fit the pattern since there is no high blood pressure and furthermore no abnormalities were found in the GCR gene and plasma CRH levels were low. It seems more probable an adaptive response to maintained low grade hypercortisolism [ 41 - 46 ].
Imagiological findings are also discordant. In classic cases a small (5 mm) round hypodense (T1-weighted sections) lesion is found; it may be hyperdense in T2-weighted sections and may or not uptake the contrast [ 15 , 16 , 22 , 47 , 48 ]. Instead in this case a large (19 × 12 mm) lesion that seems to include all the pituitary with pituitary stalk widening, no clearly visible neurohypophysis bright spot and hypointense in both T1 and T2 is found with homogenous contrast uptake. This might suggest either corticotroph hyperplasia (although evidence for CRH oversecretion is lacking as noted), silent corticotroph macroadenomas (but there is clear analytical evidence for hypercortisolism) or mixed type corticotroph adenomas (but no evidence for other hormone secretion is found) [ 26 - 28 , 30 ]. Most interestingly is the pituitary stalk widening; this finding has been reported in relation to granulomatous or immune-mediated pituitary diseases in regard with hypopituitary states [ 15 , 16 ]. Given the global involvement of the pituitary it may instead in this case correspond to the enlargement of the pars tuberalis that surrounds the pituitary stalk.
Selective removal of the pituitary adenoma whenever possible is the only curative option in CD even if the benefits of that treatment have not been proven in mild or subclinical disease [ 16 , 21 , 24 ]. Therefore the justification for surgery in this case may be questioned on clinical grounds but it is clear given the size and suprasellar extension. Surgical mortality is between 1% and 2%, success rates are between 70% and 90% in the short term, although recurrences rates of 10-15% at 10 years are reported, and in the long term hypopituitarism may occur in up to 50% of the patients, mainly in cases of macroadenoma or repeated surgeries [ 16 , 21 , 24 , 49 ].
Surgery was however ineffective, as could be appreciated early in the postoperative period [ 16 , 24 , 50 - 52 ]. At 6 months, there is clear evidence for disease persistence after surgery with marked non-suppressible hypercortisolism. Hypercortisolism was however apparently reduced to almost half, and this would favor corticotroph hyperplasia, or partial remotion of the adenoma [ 16 ]. Furthermore there is now no apparent abnormality of the pituitary except for the persistent pituitary stalk widening, so that presumably that widening at least contributes to the sustained autonomous hypercortisolism, even if a non-visible microadenoma cannot be excluded.
Pathologic findings are also atypical; an adenoma was found with no suggestion of hyperplasia, but eosinophilic or mixed type, instead of basophilic and ACTH-producing cells. Again a mixed corticotroph adenoma, a silent corticotroph macroadenoma or an intermediate lobe corticotroph adenoma remain possible but no evidence was found for other hormone secretion, clinically or analytical, or for a different proopiomelanocortin (POMC) processing, since analytical marked hypercortisolism is found [ 15 , 16 , 24 - 28 ].
There was no clear therapeutic option in this young, childless patient with minimal clinical evidence for hypercortisolism [ 16 , 24 , 53 , 54 ]. Because of minimal clinical evidence of hypercortisolism and the desire for motherhood, pituitary surgery, adrenal surgery or radiotherapy either conventional or gamma-knife were not considered appropriate; because of minimal clinical evidence of hypercortisolism medical therapies directed to the adrenals with mitothane, methyrapone or ketoconazole were not considered appropriate also given the limited efficacy of those therapies with common side effects [ 16 , 24 , 53 - 55 ].
Medical therapy directed at the pituitary was therefore attempted. As is common in CD but unusual regarding other pituitary tumors, the octreoscan was negative; but despite this somatostatin analog therapy was attempted and unsuccessful, although the new analogs may be more selective for the somatostatin receptors type 5 and may be more effective [ 16 , 24 , 56 , 57 ]. Dopaminergic therapy was attempted with cabergoline, and this seemed to be effective for some time at least, but an escape phenomenon was apparent later. Interestingly enough dopaminergic therapy has been reported as effective mainly in intermediate lobe corticotroph adenomas [ 16 , 24 , 58 , 59 ]. New therapies directed to intracellular mediators of corticotroph growth and function are not yet clinically available [ 60 ].
At this point we do not have a definitive diagnosis and even more important we do not have a clear therapeutic option. We encouraged the patient to become pregnant, assuming that this would occur spontaneously, and that CD would not complicate pregnancy given the minimal clinical features, so that pituitary surgery could be again considered, that if directed to the stalk widening may later compromise fertility. Besides a variant of CD, we cannot exclude with certainty pituitary hyperplasia secondary to CRH oversecretion, but hyperplasia was not reported and eosinophil and mixed cells were found and CRH levels were normal/low, nor even less probably cortisol resistance even if high blood pressure is lacking and weight gain is present, no abnormalities in the GCR gene were found and again CRH levels were suppressed.
Learning points
In short we report one case of what seems a common variant of CD with several worth noting characteristics: 1) female gender and young age; 2) minimal clinical features of hypercortisolism; 3) evident analytical hypercortisolism albeit with maintained cortisol circadian rhythm; 4) no evidence for increased central CRH drive; 4) macroadenoma with marked pituitary stalk widening; 5) mixed eosinophilic and clear cell adenoma; 6) persistence of the disease after apparent a successful surgery; 7) partial and only temporary response to dopaminergic agents, and no response to the somatostatin analog with absent somatostatin receptors in the octreoscan; 8) furthermore, a certain degree of acquired adaptive cortisol resistance is evident, which explains lack or only mild clinical features and therefore the rarity of this clinical presentation.
We assume many other patients like this one have been found and recognized by other centers and we strongly encourage a specific variant of CD to be thus defined.
Acknowledgments
None to declare.
Financial Disclosure
Conflict of Interest
Informed Consent
Written informed consent was obtained from the patient ABS regarding publication of this report including figures.
Author Contributions
JMM assisted the patient and wrote the manuscript; CMM thoroughly discussed the case and co-wrote the manuscript; JM thoroughly discussed the case, performed surgery and thoroughly discussed the manuscript; DLP thoroughly discussed the case, performed pathologic examination and thoroughly discussed the manuscript.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
| References |
- Broersen LH, Pereira AM, Jorgensen JO, Dekkers OM. Adrenal insufficiency in corticosteroids use: systematic review and meta-analysis. J Clin Endocrinol Metab. 2015;100(6):2171-2180. doi pubmed
- Ejaz S, Vassilopoulou-Sellin R, Busaidy NL, Hu MI, Waguespack SG, Jimenez C, Ying AK, et al. Cushing syndrome secondary to ectopic adrenocorticotropic hormone secretion: the University of Texas MD Anderson Cancer Center Experience. Cancer. 2011;117(19):4381-4389. doi pubmed
- Zhang C, Jin J, Xie J, Ye L, Su T, Jiang L, Zhou W, et al. The clinical features and molecular mechanisms of ACTH-secreting pancreatic neuroendocrine tumors. J Clin Endocrinol Metab. 2020;105(11):3449-3458. doi pubmed
- Rossi R, Tauchmanova L, Luciano A, Di Martino M, Battista C, Del Viscovo L, Nuzzo V, et al. Subclinical Cushing's syndrome in patients with adrenal incidentaloma: clinical and biochemical features. J Clin Endocrinol Metab. 2000;85(4):1440-1448. doi pubmed
- Terzolo M, Reimondo G, Chiodini I, Castello R, Giordano R, Ciccarelli E, Limone P, et al. Screening of Cushing's syndrome in outpatients with type 2 diabetes: results of a prospective multicentric study in Italy. J Clin Endocrinol Metab. 2012;97(10):3467-3475. doi pubmed
- Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res. 2004;27(3):193-202. doi pubmed
- Chiodini I, Mascia ML, Muscarella S, Battista C, Minisola S, Arosio M, Santini SA, et al. Subclinical hypercortisolism among outpatients referred for osteoporosis. Ann Intern Med. 2007;147(8):541-548. doi pubmed
- Glintborg D, Henriksen JE, Andersen M, Hagen C, Hangaard J, Rasmussen PE, Schousboe K, et al. Prevalence of endocrine diseases and abnormal glucose tolerance tests in 340 Caucasian premenopausal women with hirsutism as the referral diagnosis. Fertil Steril. 2004;82(6):1570-1579. doi pubmed
- Leon-Justel A, Madrazo-Atutxa A, Alvarez-Rios AI, Infantes-Fontan R, Garcia-Arnes JA, Lillo-Munoz JA, Aulinas A, et al. A probabilistic model for Cushing's syndrome screening in at-risk populations: a prospective multicenter study. J Clin Endocrinol Metab. 2016;101(10):3747-3754. doi pubmed
- Lindholm J, Juul S, Jorgensen JO, Astrup J, Bjerre P, Feldt-Rasmussen U, Hagen C, et al. Incidence and late prognosis of cushing's syndrome: a population-based study. J Clin Endocrinol Metab. 2001;86(1):117-123. doi pubmed
- Stewart PM, Newell-Price JDC. Chapter 15. The adrenal cortex. In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM (eds). Williams Textbook of Endocrinology. 13th edition. Elsevier, Philadelphia. 2016. p. 490-555. doi
- Juszczak A, Morris DG, Grossman AB, Nieman LK. Chapter 13. Cushing's syndrome. In: Jameson JL, De Groot LJ, Kretser DM, Giudice LC, Grossman AB, Melmed S, Potts JT, et al. (eds). Endocrinology: Adult & Pediatric, 7th edition. Elsevier Saunders, Philadelphia; 2016. p. 227-255. doi
- Rubinstein G, Osswald A, Hoster E, Losa M, Elenkova A, Zacharieva S, Machado MC, et al. Time do diagnosis in Cushing's syndrome: a meta-analysis based on 5367 patients. J Clin Endocrinol Metab. 2020;105(3):e12-e23. doi pubmed
- Papakokkinou E, Olsson DS, Chantzichristos D, Dahlqvist P, Segerstedt E, Olsson T, Petersson M, et al. Excess morbidity persists in patients with Cushing's disease during long-term remission: a Swedish nationwide study. J Clin Endocrinol. 2020;105(8):2616-2624. doi pubmed
- Melmed S, Kleinberg D. Chapter 9. Pituitary masses and tumors. In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM (eds). Williams Textbook of Endocrinology. 13th edition. Elsevier, Philadelphia; 2016. p. 232-299. doi
- Bertagna X, Guignat L, Raux-Demay M-C, Guilhaume B, Girard F. Chapter 16. Cushing's disease. In: Melmed S (ed). The Pituitary, third edition. Elsevier Academic Press, London; 2011. p. 533-617. doi
- Gicquel C, Le Bouc Y, Luton JP, Girard F, Bertagna X. Monoclonality of corticotroph macroadenomas in Cushing's disease. J Clin Endocrinol Metab. 1992;75(2):472-475. doi pubmed
- Theodoropoulou M, Reincke M, Fassnacht M, Komada M. Decoding the genetic basis of Cushing's disease: USP8 in the spotlight. Eur J Endocrinol. 2015;173(4):M73-83. doi pubmed
- Cassarino MF, Sesta A, Pagliardini L, Losa M, Lasio G, Cavagnini F, Pecori Giraldi F. Proopiomelanocortin, glucocorticoid, and CRH receptor expression in human ACTH-secreting pituitary adenomas. Endocrine. 2017;55(3):853-860. doi pubmed
- Chasseloup F, Pankratz N, Lane J, Faucz FR, Keil MF, Chittiboina P, Kay DM, et al. Germline CDKN1B loss-of-function variants cause pediatric Cushing's disease with or without an MEN4 phenotype. J Clin Endocrinol Metab. 2020;105(6):1983-2005. doi pubmed
- Bertagna X. MANAGEMENT OF ENDOCRINE DISEASE: Can we cure Cushing's disease? A personal view. Eur J Endocrinol. 2018;178(5):R183-R200. doi pubmed
- Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, Montori VM. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93(5):1526-1540. doi pubmed
- Galm BP, Qiao N, Klibanski A, Biller BMK, Tritos NA. Accuracy of laboratory tests for the diagnosis of Cushing syndrome. J Clin Endocrinol Metab. 2020;105(6):2081-2094. doi pubmed
- Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, Tabarin A, et al. Treatment of Cushing's syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2015;100(8):2807-2831. doi pubmed
- Meinardi JR, Wolffenbuttel BH, Dullaart RP. Cyclic Cushing's syndrome: a clinical challenge. Eur J Endocrinol. 2007;157(3):245-254. doi pubmed
- McNicol AM, Teasdale GM, Beastall GH. A study of corticotroph adenomas in Cushing's disease: no evidence of intermediate lobe origin. Clin Endocrinol (Oxf). 1986;24(6):715-722. doi pubmed
- Rajendran R, Naik S, Sandeman DD, Nasruddin AB. Pasireotide therapy in a rare and unusual case of plurihormonal pituitary macroadenoma. Endocrinol Diabetes Metab Case Rep. 2013;2013:130026. doi pubmed
- Petrossians P, Ronci N, Valdes Socin H, Kalife A, Stevenaert A, Bloch B, Tabarin A, et al. ACTH silent adenoma shrinking under cabergoline. Eur J Endocrinol. 2001;144(1):51-57. doi pubmed
- Heaney AP. Clinical review: Pituitary carcinoma: difficult diagnosis and treatment. J Clin Endocrinol Metab. 2011;96(12):3649-3660. doi pubmed
- Karageorgiadis AS, Papadakis GZ, Biro J, Keil MF, Lyssikatos C, Quezado MM, Merino M, et al. Ectopic adrenocorticotropic hormone and corticotropin-releasing hormone co-secreting tumors in children and adolescents causing cushing syndrome: a diagnostic dilemma and how to solve it. J Clin Endocrinol Metab. 2015;100(1):141-148. doi pubmed
- Barber TM, Adams E, Ansorge O, Byrne JV, Karavitaki N, Wass JA. Nelson's syndrome. Eur J Endocrinol. 2010;163(4):495-507. doi pubmed
- Haskett RF. Diagnostic categorization of psychiatric disturbance in Cushing's syndrome. Am J Psychiatry. 1985;142(8):911-916. doi pubmed
- Tirosh A, RaviPrakash H, Papadakis GZ, Tatsi C, Belyavskaya E, Charalampos L, Lodish MB, et al. Computerized analysis of brain MRI parameter dynamics in young patients with Cushing syndrome - A case-control study. J Clin Endocrinol Metab. 2020;105(5):e2069-e2077. doi pubmed
- Alwani RA, Schmit Jongbloed LW, de Jong FH, van der Lely AJ, de Herder WW, Feelders RA. Differentiating between Cushing's disease and pseudo-Cushing's syndrome: comparison of four tests. Eur J Endocrinol. 2014;170(4):477-486. doi pubmed
- Isidori AM, Kaltsas GA, Mohammed S, Morris DG, Jenkins P, Chew SL, Monson JP, et al. Discriminatory value of the low-dose dexamethasone suppression test in establishing the diagnosis and differential diagnosis of Cushing's syndrome. J Clin Endocrinol Metab. 2003;88(11):5299-5306. doi pubmed
- Salgado LR, Fragoso MC, Knoepfelmacher M, Machado MC, Domenice S, Pereira MA, de Mendonca BB. Ectopic ACTH syndrome: our experience with 25 cases. Eur J Endocrinol. 2006;155(5):725-733. doi pubmed
- Frete C, Corcuff J-B, Kuhn E, Salenave S, Gaye D, Young J, Chanson P, et al. Non-invasive diagnostic strategy in ACTH-dependent Cushing's syndrome. J Clin Endocrinol Metab. 2020;105(10):3273-3284. doi pubmed
- Newell-Price J, Morris DG, Drake WM, Korbonits M, Monson JP, Besser GM, Grossman AB. Optimal response criteria for the human CRH test in the differential diagnosis of ACTH-dependent Cushing's syndrome. J Clin Endocrinol Metab. 2002;87(4):1640-1645. doi pubmed
- Peters CJ, Storr HL, Grossman AB, Savage MO. The role of corticotrophin-releasing hormone in the diagnosis of Cushing's syndrome. Eur J Endocrinol. 2006;155:S93-S98. doi
- Ceccato F, Tizianel I, Vedolin CK, Boscaro M, Barbot M, Scaroni C. Human corticotropin-releasing hormone tests: 10 years of real-life experience in pituitary and adrenal disease. J Clin Endocrinol Metab. 2020;105(11):e3938-e3949. doi pubmed
- Chrousos GP, Vingerhoeds A, Brandon D, Eil C, Pugeat M, DeVroede M, Loriaux DL, et al. Primary cortisol resistance in man. A glucocorticoid receptor-mediated disease. J Clin Invest. 1982;69(6):1261-1269. doi pubmed
- Raef H, Baitei EY, Zou M, Shi Y. Genotype-phenotype correlation in a family with primary cortisol resistance: possible modulating effect of the ER22/23EK polymorphism. Eur J Endocrinol. 2008;158(4):577-582. doi pubmed
- Martins JM, Kastin AJ, Banks WA. Unidirectional specific and modulated brain to blood transport of corticotropin-releasing hormone. Neuroendocrinology. 1996;63(4):338-348. doi pubmed
- Martins JM, Banks WA, Kastin AJ. Acute modulation of active carrier-mediated brain-to-blood transport of corticotropin-releasing hormone. Am J Physiol. 1997;272(2 Pt 1):E312-319. doi pubmed
- Martins JM, Banks WA, Kastin AJ. Transport of CRH from mouse brain directly affects peripheral production of β-endorphin by the spleen. Am J Physiol. 1997;273:1083-1089. doi pubmed
- Martin Martins J, Vale S, Ferreira F, Fagundes MJ, Carmo I, Saldanha C, Martins ESJ. Plasma corticotropin releasing hormone during the feeling of induced emotions. Neuro Endocrinol Lett. 2010;31(2):250-255.
- Bonneville JF. Magnetic resonance imaging of pituitary tumors. Front Horm Res. 2016;45:97-120. doi pubmed
- Dogansen SC, Yalin GY, Tanrikulu S, Tekin S, Nizam N, Bilgic B, Sencer S, et al. Clinicopathological significance of baseline T2-weighted signal intensity in functional pituitary adenomas. Pituitary. 2018;21(4):347-354. doi pubmed
- Yamada S, Inoshita N, Fukuhara N, Yamaguchi-Okada M, Nishioka H, Takeshita A, Suzuki H, et al. Therapeutic outcomes in patients undergoing surgery after diagnosis of Cushing's disease: A single-center study. Endocr J. 2015;62(12):1115-1125. doi pubmed
- Ioachimescu AG. Prognostic factors of long-term remission after surgical treatment of Cushing's disease. Endocrinol Metab Clin North Am. 2018;47(2):335-347. doi pubmed
- Witek P, Zielinski G, Szamotulska K, Maksymowicz M, Kaminski G. Clinicopathological predictive factors in the early remission of corticotroph pituitary macroadenomas in a tertiary referral centre. Eur J Endocrinol. 2016;174(4):539-549. doi pubmed
- Ironside N, Chatain G, Asuzu D, Benzo S, Lodish M, Sharma S, Nieman L, et al. Earlier post-operative hypocortisolemia may predict durable remission from Cushing's disease. Eur J Endocrinol. 2018;178(3):255-263. doi pubmed
- Espinosa-de-Los-Monteros AL, Sosa-Eroza E, Espinosa E, Mendoza V, Arreola R, Mercado M. Long-term outcome of the different treatment alternatives for recurrent and persistent Cushing disease. Endocr Pract. 2017;23(7):759-767. doi pubmed
- Geer EB, Shafiq I, Gordon MB, Bonert V, Ayala A, Swerdloff RS, Katznelson L, et al. Biochemical control during long-term follow-up of 230 adult patients with cushing disease: a multicenter retrospective study. Endocr Pract. 2017;23(8):962-970. doi pubmed
- Verhelst JA, Trainer PJ, Howlett TA, Perry L, Rees LH, Grossman AB, Wass JA, et al. Short and long-term responses to metyrapone in the medical management of 91 patients with Cushing's syndrome. Clin Endocrinol (Oxf). 1991;35(2):169-178. doi pubmed
- Pedroncelli AM. Medical treatment of Cushing's disease: somatostatin analogues and pasireotide. Neuroendocrinology. 2010;92(Suppl 1):120-124. doi pubmed
- Petersenn S, Salgado LR, Schopohl J, Portocarrero-Ortiz L, Arnaldi G, Lacroix A, Scaroni C, et al. Long-term treatment of Cushing's disease with pasireotide: 5-year results from an open-label extension study of a Phase III trial. Endocrine. 2017;57(1):156-165. doi pubmed
- Nakhleh A, Saiegh L, Reut M, Ahmad MS, Pearl IW, Shechner C. Cabergoline treatment for recurrent Cushing's disease during pregnancy. Hormones (Athens). 2016;15(3):453-458. doi pubmed
- Ferriere A, Cortet C, Chanson P, Delemer B, Caron P, Chabre O, Reznik Y, et al. Cabergoline for Cushing's disease: a large retrospective multicenter study. Eur J Endocrinol. 2017;176(3):305-314. doi pubmed
- Zhang D, Damoiseaux R, Babayan L, Rivera-Meza EK, Yang Y, Bergsneider M, Wang MB, et al. Targeting Corticotroph HDAC and PI3-Kinase in Cushing disease. J Clin Endocrinol Metab. 2021;106(1):e232-e246. doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited. Journal of Endocrinology and Metabolism is published by Elmer Press Inc.
Largest-ever analysis of its kind finds Cushing’s syndrome triples risk of deathHeart disease and infections rank as top causes of death among those with rare disorder About Endocrine Society Endocrinologists are at the core of solving the most pressing health problems of our time, from diabetes and obesity to infertility, bone health, and hormone-related cancers. The Endocrine Society is the world’s oldest and largest organization of scientists devoted to hormone research and physicians who care for people with hormone-related conditions. The Society has more than 18,000 members, including scientists, physicians, educators, nurses, and students in 122 countries. To learn more about the Society and the field of endocrinology, visit our site at www.endocrine.org . Follow us on X (formerly Twitter) at @TheEndoSociety and @EndoMedia . Colleen Williams Senior Communications Manager, Public Relations Phone: (202)-971-3611 [email protected] Jenni Glenn Gingery Director, Communications and Media Relations Phone: (202)-971-3655 [email protected] All News & Advocacy Endocrine News Podcast The Endocrine News podcast brings you the latest research and clinical advances from experts in the field, whether you are in your car, office, or out for a run. Endocrine Society Journals Our top-ranked peer-reviewed journals are among the first to publish major developments and discovery milestones. For 100 years, the Endocrine Society has been at the forefront of hormone science and public health. Read about our history and how we continue to serve the endocrine community. IntroductionCase report, statement of ethics, disclosure statement, a case report of cushing’s disease presenting as hair loss.
Emily G. Lefkowitz , Jack P. Cossman , John B. Fournier; A Case Report of Cushing’s Disease Presenting as Hair Loss. Case Rep Dermatol 28 April 2017; 9 (1): 45–50. https://doi.org/10.1159/000457898 Download citation file:
Cushing’s syndrome is a rare endocrine disorder that comprises a large group of signs and symptoms resulting from chronic exposure to excess corticosteroids. Most cases of Cushing’s syndrome are due to increased adrenocorticotropic hormone production from a pituitary adenoma, which is referred to as Cushing’s disease. Most of the signs and symptoms are nonspecific and common in the general population, making a diagnosis often challenging. However, several dermatological manifestations, such as fragile skin, easy bruising, and reddish purple striae, are more discriminatory. Because uncontrolled Cushing’s syndrome of any etiology is associated with substantial morbidity, including increased cardiovascular disease and mortality, it is important to make an early diagnosis. Unfortunately, median delays of 2 years to diagnosis have been reported. We report a case of a woman who had multiple dermatological findings, including facial plethora, easy bruising, violaceous striae, hirsutism, and acne, the latter 2 signs reflecting androgen excess. Of interest, our patient presented with a chief complaint of hair loss, a common complaint in the general population that occurs with a greater frequency in patients with Cushing’s disease and is attributed to androgenetic alopecia, but it is rarely the presenting symptom. Cushing’s syndrome is characterized by signs and symptoms that result from prolonged exposure to excessive plasma corticosteroids. While the most common cause is iatrogenic from medically prescribed corticosteroids, endogenous Cushing’s syndrome is a relatively rare disease. A recent US-based study estimated the incidence of endogenous Cushing’s syndrome at 8 per million people [ 1 ]. Pituitary-secreting tumors, first described by Harvey Cushing in 1912 and now referred to as Cushing’s disease, account for approximately 70% of cases, while ectopic adrenocorticotropic hormone (ACTH)-secreting tumors and adrenal tumors account for 10 and 20% of cases, respectively [ 2 ]. Although the diagnosis of Cushing’s syndrome can be straightforward, when several clinical findings are present, it is often challenging to make the diagnosis. None of the symptoms or signs are pathognomonic of the syndrome, and many symptoms (such as obesity, hypertension, glucose intolerance, weight gain, fatigue, weakness, menstrual abnormalities, and depression) are found in the general population. In contrast, the dermatological manifestations of Cushing’s syndrome that include skin atrophy, alopecia, easy bruisability, and striae are less commonly observed in other individuals [ 3 ]. We report an unusual case of a woman who presented with a chief complaint of hair loss and who was found to have Cushing’s disease. A 33-year-old female with a history of gastritis, depression, and obesity presented to the dermatology clinic with a chief complaint of hair loss. She reported that the hair on her head began to thin rapidly 6 months prior to presentation, although she had been experiencing progressive thinning for about 1 year. There was minimal improvement with daily minoxidil 5% foam after 3 months. The patient had a pituitary microadenoma that was known to exist for at least a decade, but a recent magnetic resonance imaging had shown the microadenoma to be stable in size. Serum prolactin, follicle-stimulating hormone, and luteinizing hormone levels were normal. Physical examination showed a middle-aged female in no acute distress with facial plethora and prominent truncal, facial, and nuchal adiposity. Examination of the skin revealed diffuse nonscarring alopecia on the frontal scalp and vertex of the scalp (Fig. 1 ), and miniaturized hairs were seen on dermoscopy. To a lesser extent, the density of temporal and occipital hair was also decreased. There were erythematous inflammatory papules and pustules on her face and mid to lower back, violaceous striae on the abdomen and axillae, and ecchymoses on the abdomen and extremities. Facial hirsutism and patches of coarse hypertrichosis on her back were also noted (Fig. 2 ).  Diffuse hair loss on the frontal scalp and vertex of the scalp with decreased thickness of temporal and occipital hair.  Coarse patches of hypertrichosis and erythematous inflammatory papules and pustules on the mid to lower back and violaceous striae on the flanks bilaterally. Serum ferritin, iron, thyroid-stimulating hormone, antinuclear antibody, albumin, total protein, and vitamin B 12 were all negative or within normal limits. Punch biopsy from the scalp showed a normal number of follicles without fibrosis or follicular dropout but a striking increase in the ratio of catagen and telogen hairs to anagen hairs and a sparse peribulbar lymphocytic infiltrate. Subsequent tests were ordered: 24-h urine cortisol: 384 µg (range 3.5–45 µg/24 h), dehydroepiandrosterone sulfate: 380 µg/dL (range 45–270 µg/dL), and ACTH: 76 pg/mL (range 9–52 pg/mL). The results were consistent with a diagnosis of Cushing’s disease. The patient was referred to an endocrinologist and subsequently underwent a transsphenoidal resection of the pituitary microadenoma. Dermatologists may encounter skin findings that reflect an underlying endocrine disorder. While many of the signs and symptoms of Cushing’s syndrome are nonspecific, those features that best distinguish Cushing’s syndrome are proximal muscle weakness, facial plethora, easy bruising, and purple (violaceous) striae [ 3 ]; the latter 3 dermatological findings were seen in our patient on physical examination. The often prominent skin findings reflect the hypercatabolic effects of hypercortisolism – inhibition of epidermal cell division and collagen synthesis, resulting in thinning of the stratum corneum and loss of subcutaneous fat [ 4 ]. Skin atrophy may be prominent, and the loss of subcutaneous connective tissue results in easy bruising after minimal injury. The atrophy and disruption of collagenous subcutaneous fibers lead to the development of broad, purple striae because the increasingly thin skin does not hide the color of venous blood in the underlying dermis. Another skin finding is hyperpigmentation due to excess ACTH, which is most commonly seen in ectopic ACTH syndrome, less commonly in Cushing’s disease (i.e., pituitary-secreting ACTH tumor), and never in adrenal Cushing’s syndrome; the hyperpigmentation is a result of ACTH binding to melanocyte-stimulating hormone receptors. In contrast to the aforementioned skin changes, female baldness or female hair loss has been variably reported and is rarely the chief complaint of a patient presenting with Cushing’s syndrome [ 2 ]. In a review of Cushing’s syndrome that included 7 case series of 33–100 patients, 4 case series did not report female baldness, whereas it was reported in 3 of the case series with an incidence ranging from 13 to 51% [ 5 ]. More recently, the European Registry on Cushing’s Syndrome reported “hair loss” in 110 of 351 (31%) patients with Cushing’s syndrome and in 76 of 224 (34%) patients in the subset with Cushing’s disease [ 6 ]. In a recent matched case-control study using a commercial health-care insurance claims database that included 1,875 patients with Cushing’s syndrome, female balding was found to be 5 times more common than in the matched controls; the combination of weakness/fatigue and female baldness was 10 times more common [ 7 ]. Female balding in Cushing’s syndrome results from androgen hypersecretion that occurs in ACTH-dependent forms (i.e., pituitary or ectopic ACTH tumors) or with adrenocortical cancers but never with adrenal adenomas. Androgenetic alopecia, often referred to as female-pattern hair loss (FPHL), is a nonscarring form of hair loss in which the growth (anagen) phase of hair follicles is shortened resulting in follicular miniaturization [ 8 ]. The frontal scalp and vertex of the scalp are the primary sites of involvement, and the occipital scalp is usually relatively spared. The exact pattern of hair loss varies among women but typically presents with a diffuse central thinning pattern or with prominent frontal scalp thinning referred to as a “Christmas tree” pattern. FPHL is a common condition with a reported prevalence of 19% in 1 series of 1,006 Caucasian women in the United States [ 9 ]. While most women show no signs of androgen excess, a careful workup is warranted. In a series of 109 women referred for the evaluation of moderate to severe FPHL, laboratory evidence for hyperandrogenism was present in 39%, with the most common diagnosis being polycystic ovarian syndrome [ 10 ]. Our patient was noted to have an elevated dehydroepiandrosterone sulfate level, a metabolic intermediate in the biosynthesis of androgens. Women who present with FPHL should be evaluated for associated signs or symptoms of androgen excess, such as hirsutism, moderate to severe acne, and irregular menses or amenorrhea. As expected, our patient who presented with alopecia had associated findings of hirsutism and acne. It should be noted, however, that in Cushing’s syndrome the amenorrhea is likely due to cortisol-mediated inhibition of gonadotrophin release rather than to hyperandrogenism. In a review of Cushing’s syndrome, Findling and Raff [ 11 ] noted that the diagnosis of Cushing’s syndrome “is the most challenging problem in clinical endocrinology.” Patients with Cushing’s syndrome and persistent hypercortisolism have a 4–5 times excess mortality compared to the general population, highlighting the urgency of diagnosis [ 12 ]. In the European Registry on Cushing’s Syndrome, the median delay to diagnosis was a remarkable 2 years [ 6 ]. They found that general practitioners were consulted 76% of the time, endocrinologists 25%, gynecologists 24%, psychiatrists/psychologists 12%, rheumatologists 11%, and dermatologists 8% of the time. As noted above, the majority of clinical signs and symptoms of Cushing’s syndrome are relatively nonspecific, whereas dermatological manifestations, such as fragile skin, easy bruising, and purple striae, are more discriminatory [ 3 ]. Broder et al. [ 7 ] noted hirsutism to be 61 times more common in Cushing’s syndrome than in the general population. In contrast, FPHL is commonly seen in the general population, although with a 5 times greater frequency in patients with Cushing’s syndrome. Our patient presented with a history of a pituitary adenoma and findings of central obesity, easy bruising, purple striae, hypertrichosis, and female balding, and a diagnosis was rapidly made. Of interest, the case was unusual in that it was the hair loss that caused the patient to seek medical attention. The authors have no ethical conflicts to disclose. The patient has given her informed consent, including the use of the photographs. The authors have no conflicts of interest to disclose. No funding support was obtained for this work.  Email alertsCiting articles via, suggested reading.
INFORMATION
SERVICES FOR
Karger International
This Feature Is Available To Subscribers OnlySign In or Create an Account Diagnostic dilemma in Cushing’s syndrome: discrepancy between patient-reported and physician-assessed manifestations
Cite this articleYou have full access to this open access article 
Early diagnosis and immediate treatment of Cushing’s syndrome (CS) are critical for a better prognosis but remain a challenge. However, few comprehensive reports have focused on this issue or investigated whether patient-reported manifestations are consistent with physician-assessed symptoms of CS. This study aimed to clarify the differences in patient-reported and physician-assessed manifestations of signs and symptoms of CS that prevent early diagnosis. This single-center retrospective study included 52 patients with CS (16 with Cushing’s disease and 36 with adrenal CS). Upon clinical diagnosis, medical records were used to independently review the patient-reported and physician-assessed manifestations of typical (such as purple striae and proximal myopathy) and nonspecific features (such as hirsutism and hypertension). The correlations and differences between the patient-reported and physician-assessed manifestations were then analyzed. We observed a positive correlation between the total number of manifestations of nonspecific features reported by patients and those assessed by physicians, but not for typical features. Moreover, manifestations reported by the patients were less frequent than those assessed by physicians for typical features, leading to discrepancies between the two groups. In contrast, there were no differences in most nonspecific features between the patient-reported and physician-assessed manifestations. Notably, the concordance between patient-reported and physician-assessed manifestations of typical features was not associated with urinary free cortisol levels. Regardless of disease severity, patients often do not complain of the typical features of CS that are crucial for formulating a diagnosis. Avoid common mistakes on your manuscript. IntroductionEndogenous Cushing’s syndrome (CS) is caused by chronic and excessive glucocorticoid exposure. This occurs primarily due to adrenocorticotropic hormone (ACTH)-producing pituitary tumors (Cushing’s disease; CD) or cortisol-producing adrenal tumors (adrenal Cushing’s syndrome; ACS) [ 1 ]—and has a high mortality rate owing to cardiovascular disease, severe infection, and suicide, even when diagnosed and treated appropriately [ 1 , 2 ]. Moreover, the prognosis is poor if the disease is not adequately treated or remains undiagnosed [ 2 ]. Therefore, early diagnosis and immediate intervention are important, as remission of CS due to surgical and pharmacological treatment can reduce the risk of mortality [ 3 , 4 ]. CS is a rare disease with a prevalence of 57 per million individuals and an annual incidence of 3.2 per million, and its epidemiology is consistent across various regions worldwide [ 5 , 6 ]. Most symptoms and signs of CS are common in general metabolic disorders, including obesity, hypertension, osteoporosis, and diabetes mellitus [ 7 ]. However, CS should be suspected if these symptoms appear as unusual features for their age [ 1 , 8 ]. Consequently, the identification of CS is challenging and labor-intensive [ 1 , 9 , 10 ]. In fact, recent research revealed that a definitive diagnosis of CD (the most common form of CS), took an average of 3.8 ± 4.8 years from the onset of symptoms, and patients typically consulted 4.6 ± 3.8 medical professionals before this disease was identified [ 11 ]. Typical features of CS include symptoms of moon face, central obesity, or buffalo hump [ 12 ], which are similar to other symptoms such as primary obesity and therefore can lead to misdiagnosis. Furthermore, although purple striae or thin skin with an increased propensity for bruising are other typical features of CS [ 12 ], these attributes are not commonly acknowledged by the general population [ 1 , 9 ]. Attempts have been made to diagnose CS early, including the development of scoring systems to estimate the pre-test probability of CS and facial image analysis software to diagnose the specific facial features of CS [ 13 , 14 , 15 ]; however, these have not yet been used widespread or fully and the early diagnosis of CS remains dependent on the experience-based medical skills of the clinical staffs [ 16 ]. Additionally, although it is difficult for patients to recognize complex and nonspecific symptoms [ 17 , 18 ], the significance of patients recognizing their illness has recently been reported for various diseases such as heart failure and malignant carcinoma [ 19 , 20 , 21 ]. It is widely acknowledged that patients’ self-recognition can result in early detection of the disease, reduce its severity and recurrence, and enhance their quality of life [ 19 ]. In patients with endocrine diseases, there is increasing focus on issues surrounding self-recognition [ 22 , 23 , 24 ]. For example, a previous study focusing on acromegaly reported a discrepancy between patient-reported and physician-reported manifestations and indicated that resolving this discrepancy could shorten the time to diagnosis [ 25 ]. Identifying CS may be challenging for primary care physicians who are yet to specialize. Therefore, endocrinologists with extensive experience in CS have often noticed that patients and these physicians struggle to identify the symptoms of CS; however, few comprehensive reports have focused on this issue or investigated whether patient-reported manifestations are consistent with physician-assessed symptoms of CS. Therefore, this study aimed to investigate the unreported manifestations of CS among individuals referred to non-specialist healthcare providers, including primary care physicians, and to recognize potential challenges with the current diagnosis of CS with the goal of facilitating early detection. Materials and methodsPatients, study design, and data collection. This single-center retrospective study was conducted to identify the discrepancies between patient-reported and physician-assessed symptoms and investigate the factors causing these differences. From September 2004 to December 2022, 199 patients were referred to our department at a tertiary medical institution upon suspicion, evaluation, or follow-up for hypercortisolism. Of these patients, 92 were newly diagnosed with CS (36 with CD, 51 with ACS, and 5 with ectopic ACTH syndrome) based on the diagnostic guidelines [ 3 , 8 , 12 ], with a diagnosis confirmed by pathological evaluation after surgical resection [ 26 ]. However, 35 patients were excluded due to a lack of detailed clinical data on the manifestations at diagnosis. Similarly, we excluded individuals diagnosed with ectopic ACTH syndrome because of the lack of comprehensive information on symptoms reported by the patients and primary care physicians due to the rapid progression and severity of this disease. Therefore, 52 patients (16 with CD and 36 with ACS) were enrolled in this study. Upon clinical diagnosis, the manifestations included in the comprehensive standardized interview at the time of diagnosis and those assessed by the physician through collaborative assessment with multiple board-certified endocrinologists as routine practice were independently reviewed from the medical records. We categorized these manifestations reviewed from the medical records into the following two categories based on the diagnostic guidelines including those of the Japan Endocrine Society: typical features, including moon face, central obesity or buffalo hump, purple striae of ≥1 cm, thin skin and easy bruising, and proximal myopathy; and nonspecific features (shown as atypical in Japan Endocrine Society’s guideline), including hypertension, menstrual abnormalities, acne, hirsutism, peripheral edema, glucose metabolism impairment, osteoporosis, pigmentation (which is not expected in patients with ACS), and mental abnormalities [ 1 , 8 , 12 ]. Central obesity or buffalo hump can also be observed in pseudo CS. However, in this study, features were classified as the same typical feature according to clinical guidelines [ 12 , 27 ]. We also reviewed the biochemical findings, comorbidities, duration from the initial recognition of CS-related symptoms to diagnosis, and number of medical institutions visited before diagnosis. The present retrospective study was performed in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Kobe University Hospital (Approval No. 1351). The patients had the option of an opt-out process, and all procedures were part of routine medical care. Definition of patient-reported and physician-assessed manifestationsIn the context of routine clinical care, physicians asked the patients about the presence or absence of manifestations and comorbidities (e.g., hypertension, menstrual abnormalities, glucose metabolism impairment, osteoporosis, and mental abnormalities), which were documented in the medical records. These reports in the medical records were defined as patient-reported manifestations in this study. In contrast, the manifestations and comorbidities of CS were assessed within several weeks after the patient was referred to our department for suspected CS. Additional diagnostic information on comorbidities is provided in the subsequent section. Physician-assessed manifestations were subsequently defined based on these findings. Comorbidities of Cushing’s syndromeAll comorbidities were diagnosed according to the appropriate guidelines [ 28 , 29 , 30 ]. For example, hypertension was diagnosed if patients were taking oral antihypertensive medication or had more than grade 1 hypertension (≥140/90 mmHg) in a treatment-naïve state [ 28 ]. Moreover, glucose metabolism impairment—including diabetes mellitus, impaired glucose tolerance, and impaired fasting glucose—was diagnosed based on the results of blood glucose levels during fasting and after a 75-g oral glucose tolerance test, as well as hemoglobin A1c (HbA1c) levels [ 29 ]. Patients taking medications for diabetes mellitus at the time of CS diagnosis were also categorized as having diabetes. Other comorbidities included mental abnormalities, menstrual abnormalities, and the presence of osteoporosis. Mental abnormalities were defined as the use of anxiolytic medications, sleeping pills, or antidepressants prescribed by experienced psychologists, and menstrual abnormalities were defined as women with irregular menstrual cycles. Furthermore, the presence of osteoporosis was defined as bone mineral density (BMD) of <–2.5 standard deviations (SD) of the T-score of the lumbar vertebrae (L2–L4), femoral neck, or distal radius measured using dual-energy x-ray absorptiometry (DXA; Horizon A DXA System), and/or an experience of a fragility fracture [ 30 ]. As per the specifications of the measurement system employed, L1 was not included in the assessment. The Z-score was also employed as a diagnostic reference among young adults. Patients also diagnosed with osteoporosis who were receiving medications for this disease. Hormone assayIn this study, blood samples were collected after an overnight fast. Subsequently, serum cortisol levels were measured using a chemiluminescent enzyme immunoassay [CLEIA] (TOSOH, Tokyo, Japan, RRID:AB_3099658) or enzyme immunoassay [EIA] (TOSOH, Tokyo, Japan, RRID:AB_3076600). Similarly, plasma ACTH levels were measured using a CLEIA (TOSOH, Tokyo, Japan, RRID:AB_3099657, or Siemens, Tokyo, Japan, RRID:AB_2909441) and EIA (TOSOH, Tokyo, Japan, RRID:AB_2783633). In both methods, the measurements showed good correlation and no conversion was required [ 31 , 32 ]. Urinary free cortisol (UFC) levels were also measured using radioimmunoassays (RIA; TFB, Tokyo, Japan, RRID:AB_2894408) or chemiluminescent immunoassays (CLIA; Siemens, Tokyo, Japan, RRID:AB_2893154). Using the following formula, the UFC levels measured by RIA were then corrected to the value measured by CLIA: Y = 0.832X − 4.23 (Y = UFC levels using CLIA, X = UFC levels using RIA) [ 33 ]. Statistical analysisAll statistical analyses were performed using SPSS ver. 28.0 software (IBM Corp., Armonk, NY, USA). All continuous variables were analyzed using the Shapiro–Wilk normality test to confirm a normal distribution, whereas Fisher’s exact test was used to analyze categorical data. Between the two groups, differences in normally or non-normally distributed data were compared using the unpaired Student’s t -test or the Mann–Whitney U test, respectively. Cohen’s kappa coefficient was used to describe the concordance between the patient-reported and physician-assessed manifestations. As previously reported [ 19 , 20 , 34 ], the concordance based on the value of Cohen’s kappa coefficient was rated as follows: 0.00–0.20 for “Slight,” 0.21–0.40 for “Fair,” 0.41–0.60 for “Moderate,” 0.61–0.80 for “Substantial,” and 0.81–1.00 for “Almost Perfect.” For correlation analysis between two variables of non-normally distributed data, we used Spearman’s rank correlation coefficient. Multivariate logistic regression analyses were then performed to investigate variables associated with the discrepancies between patient-reported and physician-assessed manifestations. The results are presented as mean ± SD for normally distributed data and median [interquartile range] for non-normally distributed data, and differences were considered statistically significant when the P value was <0.05. Clinical characteristics of the patientsWe included 52 patients diagnosed with CS in this study. Their clinical characteristics are presented in Table 1 . Notably, this group consisted of 5 males and 47 females, with a mean age of 49.4 ± 15.8 years, median body mass index (BMI) of 23.0 [21.3–28.0] kg/m 2 , and median UFC level of 272.1 [126.0–435.0] µg/day. Of the CS patients, 16 had CD and 36 had ACS, which is consistent with epidemiological data on CS observed in Asians (including Japanese individuals); however, this differed from epidemiological data from Western countries [ 35 , 36 ]. Regarding comorbidities, 43 patients were diagnosed with hypertension—of which 34 were prescribed antihypertensive medications—with a mean systolic blood pressure (BP) of 136.4 ± 21.5 mmHg and diastolic BP of 83.5 ± 15.0 mmHg. In addition, 44 patients were diagnosed with glucose metabolism impairment—of which, 20 were prescribed oral hypoglycemic agents and/or insulin—with a median fasting serum glucose level of 99.5 [87.3–116.5] mg/dL and median HbA1c level of 6.3% [5.7–7.4]. Moreover, 29 patients were diagnosed with osteoporosis, of which 4 were prescribed antiosteoporosis medication, with BMD T-score SDs of -1.54 ± 1.39, -1.76 ± 1.12, and -0.50 [-1.53–0.50] for the lumber spine, femoral neck, and distal radius, respectively. Notably, the UFC levels were higher in patients with CD than in those with ACS (412.6 [243.2–1,100.3] vs . 215.3 [114.0–387.8] µg/day); however, there were no significant differences attributed to sex, age, BMI, or the proportion of patients with respect to comorbidities, including hypertension and glucose metabolism impairment, between patients with CD and ACS. The median duration from the patients’ initial recognition of CS-related manifestations to diagnosis was 44.0 [13.3–125.3] months, and it took more than 3 years to diagnose CS in 30 patients (58%). Furthermore, the median number of medical facilities visited by patients before diagnosis was 3.0 [2.0–5.0]; however, there were no significant differences in the duration or number of medical institutions between patients with CD and those with ACS. Frequency and concordance between patient-reported and physician-assessed CS-related manifestationsEach manifestation reported by a patient or assessed by a physician is shown vertically for individual cases in Fig. 1 . Compared with nonspecific features, typical features appeared to not be reported by the patients but were only assessed by the physicians. In addition, compared to nonspecific features, there were fewer cases in which the manifestations reported by the patients were consistent with those assessed by physicians for typical features.  Consistency between patient-reported and physician-assessed manifestations for each individual case. The consistencies or discrepancies between patient-reported and physician-assessed manifestations are shown. Vertical lines represent manifestations in individual patients. CD Cushing’s disease, ACS adrenal Cushing’s syndrome Consistent with the impact of these visually distinctive presentations shown in Fig. 1 , no correlation was observed in the number of typical features between patient-reported and physician-assessed manifestations ( r = –0.20, P = 0.16) (Fig. 2A ), whereas a positive correlation was found for nonspecific features ( r = 0.62, P < 0.01) (Fig. 2B ). Moreover, the total number of patient-reported manifestations of typical features was lower than that of physician-assessed manifestations (1.0 [0.0–2.0] vs . 3.5 [3.0–4.0], P < 0.01), and four of the five typical features were reported less frequently by patients than by physicians, except for proximal myopathy (Table 2A ). According to Cohen’s kappa coefficient, the concordance between patient-reported and physician-assessed manifestations was marked as “Fair” to “Slight,” indicating a discrepancy for all typical features. Similarly, the total number of patient-reported manifestations of nonspecific features was also lower than that in physicians (2.5 [2.0–3.0] vs . 4.0 [3.0–5.0], P < 0.01). However, except for glucose metabolism impairment or osteoporosis, there were no differences in the frequencies of nonspecific features between patient-reported and physician-assessed manifestations, and the concordance of the nonspecific features between the patient-reported and physician-assessed manifestations was “Almost perfect” for menstrual abnormality and “Substantial” for mental abnormality and hypertension, whereas that for glucose metabolism impairment and osteoporosis was “Fair.” This suggests that the discrepancy between patient-reported and physician-assessed manifestations was more significant for typical than for nonspecific features. However, no differences in these discrepancies were observed between patients with CD and those with ACS (Table 2B, C ).  Correlation between the total number of patient-reported and physician-assessed manifestations. Correlations between the total number of patient-reported and physician-assessed manifestations are shown for typical ( A ) and nonspecific features ( B ). CD is plotted by ×, and ACS is plotted by ○. The Spearman’s rank correlation coefficients and P value are presented. CI confidence interval, CD Cushing’s disease, ACS adrenal Cushing’s syndrome We performed logistic regression analyses using UFC to investigate whether excess cortisol levels influenced the discrepancy between patient-reported and physician-assessed manifestations. Notably, we observed no association between UFC levels and discrepancies between patient-reported and physician-assessed manifestations in the univariate or multivariate logistic regression analyses adjusted for sex and age (Table 3A ). In addition, no association was observed after adjusting for other variables such as BMI and disease duration. Similarly, we found that the serum cortisol levels after the low-dose dexamethasone suppression test (LDDST) were not associated with discrepancies between patient-reported and physician-assessed manifestations (Table 3B ). Thus, these disparities were shown to be insignificant when directly related to the severity of CS. In the present study, we highlight the challenges associated with the diagnosis of CS—a condition resulting from excessive glucocorticoid exposure—and elucidate the divergence between patient-reported and physician-assessed manifestations. Thus, this study may aid in the early detection of CS by identifying symptoms that patients are unable to recognize based on the disparities between patient-reported and physician-assessed manifestations of CS. In this study, the number of patient-reported manifestations of both typical and nonspecific features was lower than that of physician-assessed manifestations, suggesting that CS symptoms may have been overlooked by relying solely on patient reports. Additionally, analysis of the concordance between patient-reported and physician-assessed manifestations revealed a tendency for these manifestations to be inconsistent for both typical and nonspecific features, with a tendency to be more significant for typical features. Furthermore, the UFC and serum cortisol levels after the LDDST, which represent the severity of CS, were not associated with the concordance of manifestations between patients and physicians, suggesting that even in cases of severe CS, patients may not recognize their symptoms. These findings imply that typical features, which are essential for diagnosing CS, may be difficult for patients to recognize and poorly identified or conveyed to patients by non-specialist physicians, who are typically the first to interact with individuals with CS. The importance of educating healthcare providers such as primary care physicians, family physicians and gynecologists for early diagnosis of CS should be highlighted. According to a previous report on the diagnostic history of 176 patients with CD, 83% of the patients visited their family physician for manifestations such as weight gain and hypertension, while 46% visited a gynecologist for menstrual abnormalities before the diagnosis of CD [ 11 ]. Thus, the typical features of CS were not recognized. The examination may reveal nonspecific features. However, individuals who are non-specialists may not recognize these features as indications of CS. Therefore, patients are often unaware of the potential complications associated with CS. This is consistent with the results of our study, in which patient-reported and physician-assessed manifestations were more consistent for hypertension and menstrual abnormalities than for other manifestations such as typical features, glucose metabolism impairment, and osteoporosis. This makes diagnosis challenging as non-specialist physicians and, more prominently, patients may not recognize the full range of symptoms associated with CS, especially the typical features with high diagnostic value. In addition, older patients diagnosed with CS present with a lower BMI and waist circumference than younger patients [ 37 ], and they typically do not exhibit symptoms commonly associated with CS such as skin alterations, depression, hair loss, hirsutism, and reduced libido. These findings may further complicate the diagnosis of CS in elderly patients. By evaluating only the patient-reported manifestations, it appears that manifestations such as peripheral edema and proximal myopathy were more common. Possibly, these symptoms were not considered features of CS by physicians, in comparison to the degree of symptoms experienced by the patients. However, this may not necessarily imply diminishing the significance of the patient’s signs and symptoms, as these manifestations can be considered as the unidentified complaints and may result in a postponement of the diagnosis of CS. Patients may be experiencing symptoms that physicians do not perceive, indicating the importance of interview and physical examination. Further investigation is needed to elucidate underlying factors. Considering the rarity of CS, it is crucial to suspect and diagnose the condition based on clinical symptoms and perform the appropriate screening tests without over- or under-screening [ 7 ]. Although CS screening in patients with diabetes mellitus and hypertension has been reported to lead to a diagnosis in only 0–0.7% and 0.1–0.5% of these patients, respectively [ 38 , 39 , 40 , 41 ], it is ineffective in terms of false positives and cost [ 9 ]. Therefore, patients with typical features that are highly specific for CS, such as purple striae, easy bruising, and proximal myopathy [ 1 , 8 , 12 ], as well as those with obesity, diabetes mellitus, or hypertension in combination with these features, should be screened for CS [ 7 , 27 ]. However, our results suggest that these symptoms are unlikely to be self-recognized. Therefore, the appropriate screening measures must be implemented to establish an early and effective diagnosis of CS. In these situations, it is crucial for physicians to utilize their knowledge and experience to suspect CS based on symptoms such as typical features [ 10 ]. It has been reported that years of clinical experience in endocrine practice can contribute to the estimation of the pre-test probability of CS [ 16 ]. In contrast, non-specialists are less likely to encounter patients with CS in their lifetime, which can make it difficult to properly suspect CS [ 9 ]. From this perspective, it is of utmost importance that family physicians and general internists are knowledgeable regarding the manifestations that require screening for CS, as early diagnosis of this uncommon and severe condition is crucial [ 11 ]. Therefore, it is important for physicians who routinely treat patients presenting with common symptoms such as obesity, diabetes mellitus, and hypertension to meticulously interview and observe for any indicators of CS, even if the patient does not recognize them. Failure to adopt an appropriate tone in these situations may cause the disease to become undetectable. In rare disorders such as CS, in addition to enhancing public recognition of the disease, the appropriate sharing of information and provision of specialized care in clinical practice remain important issues [ 42 ]. Early identification of such rare diseases can be achieved by promoting an understanding of the disease and its symptoms among family, friends, and patients who may be the first to recognize the signs and symptoms in an individual. In fact, in a questionnaire survey of 340 patients with CS across 30 countries, the diagnosis of CS was made in 5.6% of cases by the patients themselves and in 0.9% by their family or friends [ 43 ]. In the present study, we found that it took more than 3 years to diagnose CS in 58% of the cases. If CS and its symptoms are popularized among the public, the typical features of CS could be more readily reported to physicians and the time to diagnosis might be shorter. Furthermore, a primary care physician who is well-educated and knowledgeable is crucial in ensuring that the concerns of such individuals are not overlooked. This study has some limitations. First, this single-center retrospective study included a relatively small sample size with few male patients. Second, CD and ACS have different pathologies; therefore, the frequencies of several CS-related manifestations will differ depending on their subtypes [ 3 , 44 ]. However, in this study, there was no difference in the discrepancies between patient-reported and physician-assessed manifestations in patients with CD or ACS. Nonetheless, it is crucial that comprehensive research is conducted in larger patient populations with a focus on employing methods that accurately reflect the pathophysiology of CD and ACS. Third, patient reports may be inaccurate in terms of onset and duration because they depend on the patient’s memory. Fourth, the endocrinologists who examined the patients differed, which may have affected the presence or absence of physician-assessed manifestations. Finally, this study investigated the differences between the manifestations reported by patients and those assessed by endocrinologists, although the evaluations conducted by primary care physicians, which are crucial for the early detection of CS, were not available. Future research is needed to investigate the differences in recognizing manifestations between non-specialist physicians and endocrinologists with extensive experience in CS and to examine the changes before and after education for these non-specialists to determine if they can lead to earlier diagnosis of CS. In conclusion, endocrinologists have been shown to be aware of CS-related symptoms, especially typical features, whereas patients do not recognize these manifestations, even when the disease is severe. Therefore, the key to the early diagnosis and treatment of CS is a more proactive approach of questioning and examining patients suspected of having the disease. M. Gadelha, F. Gatto, L.E. Wildemberg, M. Fleseriu, Cushing’s syndrome. Lancet 402 , 2237–2252 (2023). https://doi.org/10.1016/S0140-6736(23)01961-X Article PubMed Google Scholar P. Limumpornpetch, A.W. Morgan, A. Tiganescu, P.D. Baxter, V. Nyawira Nyaga, M. Pujades-Rodriguez, P.M. Stewart, The effect of endogenous Cushing syndrome on all-cause and cause-specific mortality. J. Clin. Endocrinol. Metab. 107 , 2377–2388 (2022). https://doi.org/10.1210/clinem/dgac265 Article PubMed PubMed Central Google Scholar M. Fleseriu, R. Auchus, I. Bancos, A. Ben-Shlomo, J. Berther, Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol 9 , 847–875 (2021). https://doi.org/10.1016/S2213-8587(21)00235-7 A. Mondin, F. Ceccato, G. Voltan, P. Mazzeo, R. Manara, L. Denaro, C. Scaroni, M. Barbot, Complications and mortality of Cushing’s disease: report on data collected over a 20-year period at a referral centre. Pituitary 26 , 551–560 (2023). https://doi.org/10.1007/s11102-023-01343-2 S. Wengander, P. Trimpou, E. Papakokkinou, O. Ragnarsson, The incidence of endogenous Cushing’s syndrome in the modern era. Clin. Endocrinol (Oxf). 91 , 263–270 (2019). https://doi.org/10.1111/cen.14014 Article CAS PubMed Google Scholar G. Giuffrida, S. Crisafulli, F. Ferraù, A. Fontana, Y. Alessi, F. Calapai, M. Ragonese, N. Luxi, S. Cannavò, G. Trifirò, Global Cushing’s disease epidemiology: a systematic review and meta-analysis of observational studies. J. Endocrinol. Invest. 45 , 1235–1246 (2022). https://doi.org/10.1007/s40618-022-01754-1 L. Giovanelli, C. Aresta, V. Favero, M. Bonomi, B. Cangiano, C. Eller-Vainicher, G. Grassi, V. Morelli, F. Pugliese, A. Falchetti, L. Gennari, A. Scillitani, L. Persani, I. Chiodini, Hidden hypercortisolism: a too frequently neglected clinical condition. J. Endocrinol. Invest. 44 , 1581–1596 (2021). https://doi.org/10.1007/s40618-020-01484-2 L.K. Nieman, B.M.K. Biller, J.W. Findling, J. Newell-Price, M.O. Savage, P.M. Stewart, V.M. Montori, The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 93 , 1526–1540 (2008). https://doi.org/10.1210/jc.2008-0125 Article CAS PubMed PubMed Central Google Scholar L.T. Braun, A. Riester, A. Oßwald-Kopp, J. Fazel, G. Rubinstein, M. Bidlingmaier, F. Beuschlein, M. Reincke, Toward a diagnostic score in Cushing’s syndrome. Front. Endocrinol. 10 , 766 (2019). https://doi.org/10.3389/fendo.2019.00766 Article Google Scholar M. Barbot, M. Zilio, C. Scaroni, Cushing’s syndrome: Overview of clinical presentation, diagnostic tools and complications. Best Pract. Res. Clin. Endocrinol. Metab. 34 , 101380 (2020). https://doi.org/10.1016/j.beem.2020.101380 I. Kreitschmann-Andermahr, T. Psaras, M. Tsiogka, D. Starz, B. Kleist, S. Siegel, M. Milian, J. Kohlmann, C. Menzel, D. Führer-Sakel, J. Honegger, U. Sure, O. Müller, M. Buchfelder, From first symptoms to final diagnosis of Cushing’s disease: experiences of 176 patients. Eur. J. Endocrinol. 172 , 285–289 (2015). https://doi.org/10.1530/EJE-14-0766 K. Kageyama, Y. Oki, S. Sakihara, T. Nigawara, K. Terui, T. Suda, Evaluation of the diagnostic criteria for Cushing’s disease in Japan. Endocr. J. 60 , 127–135 (2013). https://doi.org/10.1507/endocrj.ej12-0299 M. Parasiliti-Caprino, F. Bioletto, T. Frigerio, V. D’Angelo, F. Ceccato, F. Ferraù, R. Ferrigno, M. Minnetti, C. Scaroni, S. Cannavò, R. Pivonello, A. Isidori, F. Broglio, R. Giordano, M. Spinello, S. Grottoli, E. Arvat, A new clinical model to estimate the pre-test probability of Cushing’s syndrome: the Cushing score. Front. Endocrinol. 12 , 747549 (2021). https://doi.org/10.3389/fendo.2021.747549 A. León-Justel, A. Madrazo-Atutxa, A.I. Alvarez-Rios, R. Infantes-Fontán, J.A. Garcia-Arnés, J.A. Lillo-Muñoz, A. Aulinas, E. Urgell-Rull, M. Boronat, A. Sánchez-de-Abajo, C. Fajardo-Montañana, M. Ortuño-Alonso, I. Salinas-Vert, M.L. Granada, D.A. Cano, A. Leal-Cerro, Spanish CRISALIDA Study Group: A probabilistic model for Cushing’s syndrome screening in at-risk populations: A prospective multicenter study. J. Clin. Endocrinol. Metab. 101 , 3747–3754 (2016). https://doi.org/10.1210/jc.2016-1673 R.P. Kosilek, R. Frohner, R.P. Würtz, C.M. Berr, J. Schopohl, M. Reincke, H.J. Schneider, Diagnostic use of facial image analysis software in endocrine and genetic disorders: review, current results and future perspectives. Eur. J. Endocrinol. 173 , M39–M44 (2015). https://doi.org/10.1530/EJE-15-0429 D.E. Cipoli, E.Z. Martinez, deM. Castro, A.C. Moreira, Clinical judgment to estimate pretest probability in the diagnosis of Cushing’s syndrome under a Bayesian perspective. Arq. Bras. Endocrinol. Metab. 56 , 633–637 (2012). https://doi.org/10.1590/s0004-27302012000900006 L. Rydén, R. Sigström, J. Nilsson, V. Sundh, H. Falk Erhag, S. Kern, M. Waern, S. Östling, K. Wilhelmson, I. Skoog, Agreement between self-reports, proxy-reports and the National Patient Register regarding diagnoses of cardiovascular disorders and diabetes mellitus in a population-based sample of 80-year-olds. Age Ageing 48 , 513–518 (2019). https://doi.org/10.1093/ageing/afz033 K. Wada, H. Yatsuya, P. Ouyang, R. Otsuka, H. Mitsuhashi, S. Takefuji, K. Matsushita, K. Sugiura, Y. Hotta, H. Toyoshima, K. Tamakoshi, Self-reported medical history was generally accurate among Japanese workplace population. J. Clin. Epidemiol. 62 , 306–313 (2009). https://doi.org/10.1016/j.jclinepi.2008.04.006 D. Maeda, Y. Matsue, N. Kagiyama, K. Jujo, K. Saito, K. Kamiya, H. Saito, Y. Ogasahara, E. Maekawa, M. Konishi, T. Kitai, K. Iwata, H. Wada, M. Hiki, T. Dotare, T. Sunayama, T. Kasai, H. Nagamatsu, T. Ozawa, K. Izawa, S. Yamamoto, N. Aizawa, R. Yonezawa, K. Oka, S.-I. Momomura, T. Minamino, Inaccurate recognition of own comorbidities is associated with poor prognosis in elderly patients with heart failure. ESC Heart Fail 9 , 1351–1359 (2022). https://doi.org/10.1002/ehf2.13824 F. Ye, D.H. Moon, W.R. Carpenter, B.B. Reeve, D.S. Usinger, R.L. Green, K. Spearman, N.C. Sheets, K.A. Pearlstein, A.R. Lucero, M.R. Waddle, P.A. Godley, R.C. Chen, Comparison of patient report and medical records of comorbidities: results from a population-based cohort of patients with prostate cancer. JAMA Oncol 3 , 1035–1042 (2017). https://doi.org/10.1001/jamaoncol.2016.6744 J.M. Huerta, M.J. Tormo, J.M. Egea-Caparrós, J.B. Ortolá-Devesa, C. Navarro, Accuracy of self-reported diabetes, hypertension and hyperlipidemia in the adult Spanish population. DINO study findings. Rev. Esp. Cardiol. 62 , 143–152 (2009). https://doi.org/10.1016/s1885-5857(09)71532-4 C.D. Andela, H. Repping-Wuts, N.M.M.L. Stikkelbroeck, M.C. Pronk, J. Tiemensma, A.R. Hermus, A.A. Kaptein, A.M. Pereira, N.G.A. Kamminga, N.R. Biermasz, Enhanced self-efficacy after a self-management programme in pituitary disease: a randomized controlled trial. Eur. J. Endocrinol. 177 , 59–72 (2017). https://doi.org/10.1530/EJE-16-1015 R. Acree, C.M. Miller, B.S. Abel, N.M. Neary, K. Campbell, L.K. Nieman, Patient and provider perspectives on postsurgical recovery of Cushing syndrome. J Endocr Soc 5 , bvab109 (2021). https://doi.org/10.1210/jendso/bvab109 S.M. Webb, J. Kristensen, D. Vitali, S. van Klink, C. van Beuzekom, A. Santos, A. Nordenström, EndoERN patient survey on their perception of health care experience and of unmet needs for rare endocrine diseases. Endocrine 71 , 569–577 (2021). https://doi.org/10.1007/s12020-021-02625-0 P. Caron, T. Brue, G. Raverot, A. Tabarin, A. Cailleux, B. Delemer, P.P. Renoult, A. Houchard, F. Elaraki, P. Chanson, Signs and symptoms of acromegaly at diagnosis: the physician’s and the patient’s perspectives in the ACRO-POLIS study. Endocrine 63 , 120–129 (2019). https://doi.org/10.1007/s12020-018-1764-4 S.L. Asa, O. Mete, A. Perry, R.Y. Osamura, Overview of the 2022 WHO classification of pituitary tumors. Endocr. Pathol. 33 , 6–26 (2022). https://doi.org/10.1007/s12022-022-09703-7 M. Savas, S. Mehta, N. Agrawal, E.F.C. van Rossum, R.A. Feelders, Approach to the patient: Diagnosis of Cushing’s syndrome. J. Clin. Endocrinol. Metab. 107 , 3162–3274 (2022). https://doi.org/10.1210/clinem/dgac492 B. Williams, M. Giuseppe, S. Wilko, A.R. Enrico, A. Michel., B. Michel: 2018 ESC/ESH Guidelines for the management of arterial hypertension, https://academic.oup.com/eurheartj/article/39/33/3021/5079119 , https://doi.org/10.1093/eurheartj/ehy339 (2018). N.A. ElSayed, G. Aleppo, V.R. Aroda, R.R. Bannuru, F.M. Brown, D. Bruemmer, B.S. Collins, M.E. Hilliard, D. Isaacs, E.L. Johnson, S. Kahan, K. Khunti, J. Leon, S.K. Lyons, M.L. Perry, P. Prahalad, R.E. Pratley, J.J. Seley, R.C. Stanton, R.A. Gabbay, on behalf of the American Diabetes Association: 2. Classification and diagnosis of diabetes: standards of care in diabetes-2023. Diabetes Care 46 (Supplement 1), S19–S40 (2023). https://doi.org/10.2337/dc23-S002 T. Sözen, L. Özışık, N.Ç. Başaran, An overview and management of osteoporosis. Eur. J. Rheumatol. 4 , 46–56 (2017). https://doi.org/10.5152/eurjrheum.2016.048 Pharmaceuticals and Medical Devices Agency: Data sheet for the cortisol assay kit, https://www.info.pmda.go.jp/downfiles/ivd/PDF/480201_13E1X80174002020_A_02_04.pdf (2024), last accessed 25 May 2024. Pharmaceuticals and Medical Devices Agency: Data sheet for the ACTH assay kit, https://www.info.pmda.go.jp/downfiles/ivd/PDF/480201_23000EZX00023000_A_03_05.pdf (2024), last accessed 25 May 2024. LSI Medience Corporation: Changes to the content of clinical tests, https://www.medience.co.jp/clinical/information/parts/pdf/22-04.pdf (2022), last accessed 22 May 2024 J.R. Landis, G.G. Koch, The measurement of observer agreement for categorical data. Biometrics 33 , 159–174 (1977). https://doi.org/10.2307/2529310 S.-C. Tung, P.-W. Wang, R.-T. Liu, J.-F. Chen, C.-J. Hsieh, M.-C. Kuo, J.W. Yang, W.-C. Lee, M.-H. Cheng, T.-C. Lee, Clinical characteristics of endogenous Cushing’s syndrome at a medical center in Southern Taiwan. Int. J. Endocrinol. 2013 , 685375 (2013). https://doi.org/10.1155/2013/685375 T. Imai, H. Funahashi, Y. Tanaka, J. Tobinaga, M. Wada, T. Morita-Matsuyama, Y. Ohiso, H. Takagi, Adrenalectomy for treatment of Cushing syndrome: results in 122 patients and long-term follow-up studies. World J. Surg. 20 , 781–786 (1996). https://doi.org/10.1007/s002689900119 . V. Amodru, A. Ferriere, A. Tabarin, F. Castinetti, S. Tsagarakis, M. Toth, R.A. Feelders, S.M. Webb, M. Reincke, R. Netea-Maier, D. Kastelan, A. Elenkova, D. Maiter, O. Ragnarsson, A. Santos, E. Valassi, and for the ERCUSYN Study Group: Cushing’s syndrome in the elderly: data from the European Registry on Cushing’s syndrome. Eur. J. Endocrinol. 188 , 395–406 (2023). https://doi.org/10.1093/ejendo/lvad008 S. Budyal, S.S. Jadhav, R. Kasaliwal, H. Patt, S. Khare, V. Shivane, A.R. Lila, T. Bandgar, N.S. Shah, Is it worthwhile to screen patients with type 2 diabetes mellitus for subclinical Cushing’s syndrome? Endocr Connect 4 , 242–248 (2015). https://doi.org/10.1530/EC-15-0078 M. Terzolo, G. Reimondo, I. Chiodini, R. Castello, R. Giordano, E. Ciccarelli, Screening of Cushing’s syndrome in outpatients with type 2 diabetes: results of a prospective multicentric study in Italy. J. Clin. Endocrinol. Metab. 97 , 3467–3475 (2012). https://doi.org/10.1210/jc.2012-1323 G.H. Anderson Jr, N. Blakeman, D.H. Streeten, The effect of age on prevalence of secondary forms of hypertension in 4429 consecutively referred patients. J. Hypertens. 12 , 609–615 (1994). https://doi.org/10.1097/00004872-199405000-00015 . M. Omura, J. Saito, K. Yamaguchi, Y. Kakuta, T. Nishikawa, Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens. Res. 27 , 193–202 (2004). https://doi.org/10.1291/hypres.27.193 H. Rahabi, M. Givony, B. Demaret, F. Albarel, M.-R. Aubron, B. Bartès, L. Bernard, H. Abdoul., N. Bouazza, P. Brun, D. Drui, V. Dujardin, C. Lançon, S. Malivoir, I. Netchine., B. Perrotin, V. Picard, R. Reynaud, M. Ribeiro, V. Tardy Guidollet., A. Victor, J. Bertherat, C. Colin, T. Brue: The experience of diagnosis announcement in rare endocrine diseases: A survey of the French FIRENDO network. Ann. Endocrinol (Paris). https://doi.org/10.1016/j.ando.2023.10.008 (2023). E. Valassi, I. Chiodini, R.A. Feelders, C.D. Andela, M. Abou-Hanna, S. Idres, A. Tabarin, Unmet needs in Cushing’s syndrome: the patients’ perspective. Endocr Connect 11 , e220027 (2022). https://doi.org/10.1530/EC-22-0027 E. Valassi, Clinical presentation and etiology of Cushing’s syndrome: Data from ERCUSYN. J. Neuroendocrinol. 34 , e13114 (2022). https://doi.org/10.1111/jne.13114 Download references AcknowledgementsWe thank all the physicians and medical assistants who were involved in this study. We are grateful to all the laboratory members for their excellent discussions and fruitful suggestions. We also thank Editage ( www.editage.jp ) for English language editing. This work was partially supported by the Japan Society for the Promotion of Science (KAKENHI, grant numbers 22K08654 (HF) and 21K08555 (GI)) and the Hyogo Science and Technology Association (HF). Open Access funding provided by Kobe University. Author informationThese authors contributed equally: Yuma Motomura, Shin Urai Authors and AffiliationsDivision of Diabetes and Endocrinology, Department of Internal Medicine, Kobe University Graduate School of Medicine, Kobe, Hyogo, 650-0017, Japan Yuma Motomura, Shin Urai, Masaaki Yamamoto, Masaki Suzuki, Naoki Yamamoto, Genzo Iguchi & Wataru Ogawa Division of Diabetes and Endocrinology, Department of Internal Medicine, Kobe University Hospital, Kobe, Hyogo, 650-0017, Japan Hironori Bando & Hidenori Fukuoka Medical Center for Student Health, Kobe University, Kobe, Hyogo, 657-8501, Japan Genzo Iguchi Department of Biosignal Pathophysiology, Kobe University Graduate School of Medicine, Kobe, Hyogo, 657-8501, Japan You can also search for this author in PubMed Google Scholar ContributionsConceptualization and methodology: Y.M., S.U., G.I., and H.F.; Data curation and investigation: Y.M. and S.U.; Resources: Y.M., S.U., M.Y., M.S., N.Y., and H.F.; Formal analysis: Y.M. and S.U.; Writing - original draft preparation and visualization: Y.M. and S.U.; Writing - review and editing: H.B., M.Y., M.S., N.Y., G.I., W.O., and H.F.; Project administration and supervision: H.F. All the authors contributed to the discussion and approved the final version of the manuscript. Corresponding authorCorrespondence to Hidenori Fukuoka . Ethics declarationsConflict of interest. The authors declare no competing interests. Ethics approvalThis study conformed to the Declaration of Helsinki guidelines and was approved by the Ethics Committee of Kobe University Hospital (Approval No. 1351). Informed consentInformed consent was obtained from all the participants using an opt-out approach. Additional informationPublisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Rights and permissionsOpen Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . Reprints and permissions About this articleMotomura, Y., Urai, S., Bando, H. et al. Diagnostic dilemma in Cushing’s syndrome: discrepancy between patient-reported and physician-assessed manifestations. Endocrine (2024). https://doi.org/10.1007/s12020-024-03935-9 Download citation Received : 29 April 2024 Accepted : 14 June 2024 Published : 25 June 2024 DOI : https://doi.org/10.1007/s12020-024-03935-9 Share this articleAnyone you share the following link with will be able to read this content: Sorry, a shareable link is not currently available for this article. Provided by the Springer Nature SharedIt content-sharing initiative
An official website of the United States government The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site. The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
Cushing’s syndrome during pregnancy - two case reportsLaurence Katznelson, Stanford University, United States Associated DataThe original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author. Cushing’s syndrome (CS) during pregnancy is a rare endocrine disorder characterized by hypercortisolism, which is significantly associated with maternal-fetal complications. Despite its rarity, CS during pregnancy may be related to a high risk of complications for both the mother and fetus.The aim of the present case study is to update the diagnostic approach to CS during pregnancy and the therapeutic strategies for this medical condition to minimize maternal-fetal complications. Here, we present two cases of CS in pregnant women, one of whom had twins. Typical clinical symptoms and signs of hypercortisolism developed at the beginning of pregnancy. The plasma cortisol diurnal rhythm of the pregnant patient was absent. CS was confirmed by cortisol and adrenocorticotropic hormone (ACTH) assessment, as well as imaging examination. We investigated the changes in the hypothalamic-pituitary-adrenal axis during normal pregnancy and the etiology, diagnosis and treatment of CS during pregnancy. Due to the associated risks of laparoscopic adrenalectomy,it is uncertain whether this treatment significantly decreases overall maternal mortality. Additional observational research and validation through randomized controlled trials (RCTs) are required. We advise that CS in pregnant women be diagnosed and treated by experienced teams in relevant departments and medical centers. 1. IntroductionCushing’s syndrome (CS) is an endocrine disorder characterized by hypercortisolism with various causes. CS during pregnancy is rare but troublesome. There are fewer than 200 reported cases of this syndrome during pregnancy in the literature ( 1 ). Gonadotropin synthesis could be inhibited by excessive glucocorticoid secretion in female patients with CS during pregnancy, resulting in disorders in ovarian and endometrial functions. They are prone to oligomenorrhea, irregular menstruation and amenorrhea, and trouble getting pregnany. Additionally, expectant individuals with Cushing’s syndrome face a notably elevated risk of experiencing severe complications throughout pregnancy. Long-term exposure to hypercortisolism may cause maternal hypertension, hypokalemia, centripetal obesity, abnormal glucose metabolism, heart failure, pulmonary edema, opportunistic infections, pathological osteoporosis, bone fracture, and even death.The primary cause of CS in pregnant women is adrenal adenoma, followed by Cushing’s disease and ectopic adrenocorticotropic hormone (ACTH) syndrome (EAS). Adrenal adenoma occurs in 15% of nonpregnant women with CS and approximately 50% of pregnant women ( 2 – 4 ). The diagnosis of CS during pregnancy is challenging. Misdiagnosis of CS is also common because it can easily be confused with preeclampsia or gestational diabetes mellitus (GDM). CS during pregnancy may result in maternal-fetal complications. Not only its manifestations, including hypertension and hyperglycemia but also the underlying causes of CS require effective treatment in pregnant women. Here, we present two cases of Cushing’s syndrome (CS) during pregnancy. One of the pregnant women with CS was expecting twins. The other pregnant woman was diagnosed with ACTH-independent CS and underwent a successful unilateral laparoscopic adrenalectomy. The objective of this study is to describe therapeutic strategies for managing CS during pregnancy to minimize both maternal and fetal complications. 2. Case report2.1. case one. A28-year-old woman presented with abdominal striae and abnormal glycemic metabolism at 31 weeks of pregnancy. She was admitted to the West China Hospital of Sichuan University for gestational diabetes mellitus (GDM) and suspected Cushing’s syndrome (CS) during pregnancy. During the physical examination, her blood pressure was measured at 132/80 mmHg. She exhibited physical characteristics such as a moon face, buffalo hump, central obesity with a body mass index (BMI) of 28.9 kg/m², abdominal purple striations over 1 cm wide, and mild lower extremity edema. Given these signs, CS was suspected, leading to a referral for laboratory and functional tests related to CS. Hormonal analyses revealed serum cortisol concentrations of 892.26 nmol/L, 759.01 nmol/L, and 929.39 nmol/L at 8:00, 16:00, and 24:00, respectively (normal reference range: 118 ~ 618 nmol/L). Morning serum ACTH was <0.1 ng/L (normal range: 5 ~ 78 ng/L). Midnight saliva cortisol concentrations on three different dates were 66.89 nmol/L, 43.12 nmol/L, and 59.95 nmol/L (normal reference range: 0-10.4 nmol/L). Additionally, 24-hour urinary free cortisol (UFC) concentrations were 723.1 ug/24h and 591.2 ug/24h on two separate occasions (reference range: 20.26-127.55 ug/24h) (see Table 1 ). A 2 mg low-dose dexamethasone suppression test (LDDST) resulted in a serum cortisol concentration of 68.5 nmol/L, and an 8 mg high-dose dexamethasone suppression test (HDDST) showed a concentration of 48.7 nmol/L.Her liver and renal functions and electrolytes were normal. The patient was diagnosed with ACTH-independent CS, and the primary diagnosis was confirmed by magnetic resonance imaging (MRI), which showed a 2.6×2.5 cm adenoma in her leftadrenal gland ( Figure 1 ). There were no abnormalities on fetal ultrasound imaging. Table 1Laboratory findings and HPA of the pregnant patient with CS on admission.
 MRI of the adrenal adenoma in a woman with CS during pregnancy. The female patient successfully underwent a unilateral laparoscopic adrenalectomy at 32 weeks of gestation. Adrenalectomy was performed under general anesthesia; during the operation, she was placed in the right lateral decubitus position. No complications developed during surgery. The pathological examination of the removed adenoma revealed adrenal cortical adenoma. This was consistent with the diagnosis of ACTH-independent CS. Three days after surgery, her morning serum cortisol concentration at 8:00 was 185 nmol/L, and she also had fatigue and anorexia. She was administered 30 mg of hydrocortisone (20 mg at 8:00 a.m. and 10 mg at 4:00 p.m.) supplementation due to relative adrenal insufficiency. She was discharged 10 days after laparoscopic adrenalectomy. Her serum cortisol concentration was 200.6 nmol/L, and her cortisol level was 46.5nmol/L after a 1mg dexamethasone suppression test (DST). Her replacement therapy was therefore stopped. A follow-up visit performed 8 weeks after discharge showed normal results. Subsequently, she delivered a healthy baby at 39 weeks gestation through normal delivery, with a birth weight of 3.6 kg. Nine months post-delivery, a 1mg DST indicated cortisol concentrations of 105 nmol/L and ACTH 1.40 ng/L (normal range: 5-78 ng/L), signifying continuous remission. 2.2. Case twoA 34-year-old G2P1 female patient visited the West China Hospital of Sichuan University, reporting abnormal blood glucose levels for the past 4 years. She had experienced a weight gain of 10 kg in the last year, along with muscle weakness and elevated blood pressure over the past 4 weeks. Four years prior, she had been diagnosed with gestational diabetes mellitus (GDM) during her first pregnancy at a local hospital. Postpartum, she maintained near-normoglycemia through dietary changes, exercise, and other lifestyle interventions. One year ago, the patient developed centripetal obesity, and her attempts at weight control through diet and exercise proved ineffective. An oral glucose tolerance test (OGTT) revealed a 1-hour blood glucose concentration of 5.37 mmol/L and a 2-hour blood glucose concentration of 10.76 mmol/L after a 75g glucose load. Subsequently, she was found to be pregnant again and received treatment for GDM. During pregnancy, the patient gradually suffered from facial and back acne, abdominal purple lines, increased body hair, and decreased hairline. Laboratory findings and HPA on admission was shown in Table 1 . Due to safety concerns, the pregnant woman declined further MRI examination. It is crucial to conduct diagnostic analyses for Cushing’s syndrome (CS) early in pregnancy. In the absence of sufficient evaluation and examination for CS, the patient underwent a cesarean section at 33 weeks of gestation, delivering two male infants who required intensive care for10 days and at-home feeding after 1 month. During a subsequent visit, a 1mg dexamethasone suppression test (DST) was conducted, revealing non-inhibition of cortisol. Enhanced abdominal computed tomography (CT) displayed a 22 mm occupation of the left adrenal branch. An MRI of the sellar regionrevealed no abnormalities in the pituitary glands. Two weeks after delivery, her blood pressure measured 160/100 mmHg, and oral antihypertensive medications proved ineffective. Consequently, she was admitted to the Department of Endocrinology and Metabolism at West China Hospital. A physical examination revealed a blood pressure of 164/110 mmHg, along with symptoms such as moon face, decreased hairline, abdominal obesity, evident neck and back acne, and abdominal purple stripes. Her serum potassium level was 2.32 mmol/L. Results from the oral glucose tolerance test (OGTT) showed blood glucose levels of 5.18 mmol/L at 0 hours and 11.94 mmol/L at 2 hours after meals. Aldosterone in the decubitus position was measured at 11.65 ng/dL (normal range: 4.5-17.5 ng/dL). The supine aldosterone/decubitus renin activity ratio (ARR) was 3.34, within the normal. ACTH<1.00 ng/L (normal range: 5-78 ng/L), cortisol circadian was absent. The 24-hour urinary free cortisol (UFC) levels were 585.9 ug/24h and 738.6 ug/24h, respectively (normal range: 20.26-127.55 ug/24h). Bone mineral density (BMD) examination showed that the average Z values of the femoral neck and total hip were -1.7 and -1.7, respectively, and the average Z value of L1-L4 was -2.8. Adrenal contrast-enhanced CT revealed a 2.7x2.5 cm soft tissue density nodule of the left adrenal gland, indicating the possibility of a left adrenal adenoma. The patient was subsequently transferred to the Department of Urology and underwent laparoscopic adrenalectomy on May 11, 2016. The operation was conducted successfully, and the postoperative pathological examination indicated a left adrenal cortical adenoma. Two days after the surgery, she was transferred back to the Department of Endocrinology and Metabolism. To address adrenal insufficiency following the surgery, glucocorticoid supplementation therapy (prednisone 10 mg in the morning and 5 mg in the afternoon) was initiated. One week later, she was discharged from the hospital with a blood pressure reading of 140/94 mmHg. Her blood potassium level was 3.6 mmol/L, and her ACTH was 1.05 ng/L, with cortisol concentrations within the normal range. After discharge, the hormone dose was adjusted and gradually reduced during outpatient follow-ups. Two months post-surgery, a re-examination revealed an ACTH level of 18.46 ng/L. Two years later, the ACTH level increased to 45.84 ng/L, and the 24-hour urinary free cortisol (UFC) was measured at 100.9 ug/24h. Additionally, an oral glucose tolerance test (OGTT) displayed fasting blood glucose at 4.62 mmol/L, 2-hour blood glucose at 10.70 mmol/L, HbA1c at 4.9%, and blood potassium at 3.67 mmol/L.The twin brothers are currently thriving and well-developed. 3. DiscussionPregnancy brings about several physiological alterations, one of which involves the activation of the maternal hypothalamic-pituitary-adrenal axis. This study explores uncommon situations and results in expectant women dealing with Cushing’s syndrome (CS). CS is a collection of syndromes arising from prolonged, excessive cortisol secretion or exogenous intake, influenced by various causes. The initial documentation of Cushing’s syndrome (CS) during pregnancy was presented in 1953 by Hunt and his colleagues ( 5 ). In the initial case series, the fetal mortality rate was noted to be 43%. Recent reports have consistently affirmed that complications affecting both the mother and the fetus are prevalent in Cushing’s syndrome (CS) during pregnancy ( 6 ). As pregnancy progresses and the pituitary glands undergo growth, there is a natural physiological increase in pituitary-adrenal axis activity with advancing gestational weeks. This results in elevated levels of ACTH, hepatic corticosteroid binding globulin (CBG), increased serum, salivary, and urinary free cortisol, as well as a lack of suppression of cortisol concentration after dexamethasone administration and placental production of ACTH ( 7 ). Hence, the elevated cortisol levels induced by pregnancy pose a challenge in diagnosing Cushing’s syndrome (CS). It is frequently misdiagnosed during pregnancy due to its resemblance to gestational diabetes mellitus (GDM). In our current study, the presence of a typical Cushing’s phenotype, elevated cortisol levels coupled with low ACTH concentrations, and the identification of adrenal adenoma images on abdominal CT collectively resulted in the diagnosis of ACTH-independent Cushing’s syndrome. This case was determined to be Cushing’s syndrome caused by adrenal cortical adenoma, the most prevalent cause of CS in pregnancy. The mechanism behind pregnancy-induced Cushing’s syndrome may involve luteinizing hormone (LH) and human chorionic gonadotropin (HCG) in pregnant women inducing the overexpression of relevant receptors (such as the chorionic gonadotropin receptor), which exerts a similar effect on the ACTH receptor, leading to adrenal hyperplasia and excessive cortisol production ( 8 ). Our patients exhibited typical symptoms and signs of Cushing’s syndrome (CS). The typical clinical manifestations of pregnancy complicated with CS resemble those in nonpregnant patients, including Cushing’s appearance such as moon face and buffalo back, weight gain centripetal obesity, water and sodium retention, and so forth. CS has repercussions for both the mother and the fetus.In fetuses, a recent review indicated a noteworthy rise in fetal mortality and complications among individuals with Cushing’s syndrome (CS). These complications encompass stillbirth, premature birth, spontaneous abortion, neonatal death, fetal intrauterine growth restriction, fetal malformation, and adrenal insufficiency ( 1 ). The placenta utilizes 11β-hydroxysteroid dehydrogenase type 2 (11-β-HSD2) enzymes to enhance the metabolism of cortisol, safeguarding the fetus from excessive cortisol levels ( 9 ). In addition to the typical Cushing’s face, the second patient presented not only abnormal glucose tolerance during pregnancy, hypertension, and osteoporosis but also hypokalemia. A review of the literature revealed that there are limited reported cases of pregnancy complicated by Cushing’s syndrome and persistent hypokalemia in China ( 10 ). The occurrence of hypokalemia might also be linked to deficiencies in 11-β-HSD2 enzymes in individuals with Cushing’s syndrome. These deficiencies result in decreased conversion of cortisol to inactive corticosteroids and an augmented binding of cortisol to corticosteroid receptors, consequently elevating potassium excretion ( 11 ). In this instance, hypokalemia was a recurring issue before surgery, and the Aldosterone-to-Renin Ratio (ARR) was employed to rule out primary aldosteronism. Blood potassium levels normalized within a week after surgery, suggesting a connection between hypokalemia and Cushing’s syndrome induced by adrenal adenoma. Given the infrequency of pregnancy complicated by Cushing’s syndrome, the existing diagnostic criteria are based on empirical observations. In this case, ACTH was lower than the lower limit of the reference value, indicating that hypercortisolism may still play a major role in the feedback inhibition of the HPA axis. The 24-hour UFC was more than 3 times higher than normal, and the circadian rhythm disappeared, which was in line with the characteristics of typical CS. Currently, there is no established diagnostic standard for pregnancy complicated by Cushing’s syndrome. Typically, a combination of endocrine laboratory tests and imaging examinations is employed to diagnose suspected cases, aiming to confirm Cushing’s syndrome and provide further clarification on its etiology. Following the International Endocrine Society guidelines for diagnosing Cushing’s syndrome, recommended diagnostic measures for pregnant women suspected of having this syndrome include the measurement of midnight serum or salivary cortisol concentrations (at least twice), 24-hour urinary free cortisol (UFC), and the 2 mg low-dose dexamethasone suppression test (LDDST) ( 12 ). Plasma ACTH concentration was used for the differential diagnosis of ACTH-dependent and ACTH-independent CS. The preferred method for screening adrenal cortical adenomas is now renal ultrasound examination. In the case of pregnant individuals with suspected pituitary tumors, the safety of using nonenhanced MRI on the fetus has not been fully established ( 13 ). There are reports indicating that individuals with suspected Cushing’s syndrome in the early stages of pregnancy received a definitive diagnosis of Cushing’s disease through the use of high-resolution MRI and bilateral inferior petrosal sinus sampling (BIPSS) ( 14 ). The management of Cushing’s syndrome during pregnancy involves both surgical and medicinal approaches. It is advisable for individuals experiencing pregnancy complicated by Cushing’s syndrome to opt for surgical intervention following the early termination of the pregnancy. Typically, laparoscopic adrenalectomy is the preferred initial treatment for those with cortisol-secreting adrenal adenomas. Medication may be considered when the lesion cannot be identified or surgery is contraindicated. Due to the potential risk of spontaneous abortion in early pregnancy post-surgery and the susceptibility of the fetus to various drugs, medication treatment becomes a consideration. Additionally, anesthesia during surgery could potentially lead to premature birth in late pregnancy. Although there have been successful reports of adrenalectomy, it is noted that the laparoscopic approach is deemed safe ( 6 ). For pregnant women diagnosed with Cushing’s syndrome in the final weeks of the third trimester, medical treatment administration and the postponement of surgery until after delivery are often recommended. As indicated in a recent account, a pregnancy case involving Cushing’s syndrome caused by adrenal cortical adenoma was effectively managed through a combination of metyrapone at 0.5 g three times a day and ketoconazole at 0.4 g twice a day ( 15 ). The patient declined surgery and opted for oral medication throughout the pregnancy, demonstrating good drug tolerance and no medication-related complications. Eventually, she successfully delivered a healthy baby boy via cesarean section with an Apgar score of 8. No drug-induced teratogenic effects were observed in the neonate. Following delivery, the adenoma was surgically resected. After a five-year follow-up, the prognosis was favorable. In our observational report, the pregnant patient persisted in preserving the fetus and declined surgical intervention for Cushing’s syndrome until a successful delivery. Two years postoperative, both the mother and baby remained in good health. 4. ConclusionIn conclusion, the timely diagnosis and treatment of Cushing’s syndrome (CS) are crucial. Both surgery and medication are viable treatment options for CS during pregnancy. Our findings indicate that pregnancy should not be considered an absolute contraindication for laparoscopic adrenalectomy; rather, this procedure can be regarded as a safe and effective treatment for pregnant women with adrenal CS. However, due to the associated risks of laparoscopic adrenalectomy, it remains unclear whether the treatment significantly reduces overall maternal mortality. Further observational studies and confirmation through randomized controlled trials (RCTs) are necessary. We recommend that CS in pregnant women be diagnosed and treated by experienced teams in relevant departments and medical centers. Data availability statementEthics statement. The studies involving humans were approved by the Ethics Committee of West China Hospital of Sichuan University (approval number 2022-840). The studies were conducted in accordance with the local legislation and institutional requirements. The patient(s)/participant(s) provided written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Author contributionsSC: Data curation, Formal analysis, Investigation, Resources, Visualization, Writing – original draft. YL: Investigation, Writing – original draft. HT: Conceptualization, Funding acquisition, Supervision, Validation, Writing – review & editing. AcknowledgmentsWe would like to thank our patient and her mother for their help and enthusiasm. We also acknowledge our clinical colleagues in the Division of Endocrinology and Metabolism of West China Hospital for their help in organizing various investigations. We are grateful to Prof. Hoffman (Andrew R. Hoffman, Division of Endocrinology, Gerontology and Metabolism, Stanford University School of Medicine) for his support and advice regarding the submission and revision of this paper. Funding StatementThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the 1·3·5 project for disciplines of excellence–Clinical Research Incubation Project, West China Hospital, Sichuan University (No.2020HXFH034) and Sichuan Province Health Commission Project (No.20PJ046). AbbreviationsACTH, Adrenocorticotropic hormone; ARR, aldosterone renin-activity ratio; BMI, Body mass index; BMD, Bone mineral density; BIPSS, bilateral inferior petrosal sinus sampling; CS, Cushing’s syndrome; CT, Computed tomography; CBG, Corticosteroid binding globulin; DST, dexamethasone suppression test; EAS, ectopic ACTH syndrome; HPA, Hypothalamic-pituitary-adrenal; HbA1c, Glycosylated hemoglobin A1c; HCG, Human chorionic gonadotropin; HSD2, Hydroxysteroid dehydrogenase type 2; HDDST, High-dose dexamethasone suppression test; IGT, Impaired glucose tolerance; LH, Luteinizing hormone; LDDST, Low-dose dexamethasone suppression test; MRI, magnetic resonance imaging; OGTT, oral glucose tolerance test; GDM, gestational diabetes mellitus; UFC, urinary free cortisol. Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher. CASE REPORT articleCase report: comprehensive follow-up of a colombian family carrying a novel men1 variant linked to a rare acth-producing pancreatic neuroendocrine carcinoma. 
Background: Multiple Endocrine Neoplasia type 1 (MEN1) is an autosomal dominant disorder marked by pathogenic variants in the MEN1 tumor suppressor gene, leading to tumors in the parathyroid glands, pancreas, and pituitary. The occurrence of ACTH-producing pancreatic neuroendocrine carcinoma is exceedingly rare in MEN1. Case presentation: This report details a Colombian family harboring a novel MEN1 variant identified through genetic screening initiated by the index case. Affected family members exhibited primary hyperparathyroidism (PHPT) symptoms from their 20s to 50s. Uniquely, the index case developed an ACTH-secreting pancreatic neuroendocrine carcinoma, a rarity in MEN1 syndromes. Proactive screening enabled the early detection of pituitary neuroendocrine tumors (PitNETs) as microadenomas in two carriers, with subsequent surgical or pharmacological intervention based on the clinical presentation. Conclusion: Our findings underscore the significance of cascade screening in facilitating the early diagnosis and individualized treatment of MEN1, contributing to better patient outcomes. Additionally, this study brings to light a novel presentation of ACTH-producing pancreatic neuroendocrine carcinoma within the MEN1 spectrum, expanding our understanding of the disease’s manifestations. IntroductionMultiple Endocrine Neoplasia type 1 (MEN1) is an autosomal dominant disorder with a global prevalence ranging from 1 to 10 cases per 100,000 individuals, showing no preference for gender. Its penetrance increases significantly with age, being reported at 50% by age 20, exceeding 95% by age 40, and nearing 100% beyond age 70 ( 1 ). The condition primarily results from loss-of-function variants in the MEN1 gene on chromosome 11q13. This gene encodes Menin, a 610-amino acid scaffold protein that plays a crucial role in various cellular processes. Menin interacts with several intracellular molecules, such as JunD, NFκB, and Smad3, implicating it in transcriptional regulation, maintaining genome stability, and controlling cell division and proliferation ( 2 ). MEN1 is characterized by the development of primary hyperparathyroidism (PHPT) in 95% of cases, pituitary neuroendocrine neoplasms (PitNENs) in 30–40%, and duodenal-pancreatic neuroendocrine tumors (DP-NETs) in 40–70% of patients ( 3 ). Additionally, the presence of carcinoids, adrenocortical tumors, and facial angiofibromas highlight the syndrome’s clinical variability. The diagnosis of MEN1 is complex, dependent on either the identification of two or more MEN1-associated tumors, finding a MEN1 tumor in a patient with a first-degree relative who has a MEN1 pathogenic variant, or uncovering an asymptomatic carrier via genetic cascade screening. Given MEN1 protracted natural progression, early-stage diagnosis presents significant challenges. This difficulty is compounded by clinical variability and the interplay of genetic and environmental factors, highlighting the crucial role of genetic testing. Such testing is instrumental in not only confirming MEN1 diagnoses but also in identifying individuals at risk, ensuring timely intervention and management ( 4 ). Here, we present a comprehensive 14-year follow-up of a Colombian family affected by a novel pathogenic variant MEN1: c.698dup, p.Met233IlefsTer4. This genetic variant impacted five family members across three generations, discovered through cascade genetic screening (CGS) initiated after identifying the index case. Notably, the index case manifested with an adrenocorticotropic hormone (ACTH)-producing pancreatic neuroendocrine carcinoma, leading to ectopic Cushing’s syndrome—a presentation not previously reported in MEN1. Materials and methodsGenetic test and analysis. Molecular analysis of the MEN1 gene was conducted at the Instituto Nacional de Cancerología, Bogotá D.C., Colombia, utilizing Sanger sequencing to analyze genomic DNA from peripheral lymphocytes of the index case and consenting asymptomatic family members. We targeted the entire coding sequence and exon-intron junctions of the MEN1 gene, amplifying nine exons with eight primer pairs. The amplified products were sequenced using the BigDye Terminator Cycle Sequencing Kit v3.1 on an Applied Biosystems/HITACHI 3500 Genetic Analyzer, USA, according to standard protocols. Electropherograms were analyzed against reference sequences NG_008929.1 and NM_130799.2 using SeqA v6.0 and SeqScape v3.0 software from Applied Biosystems, USA, ensuring precise variant identification. Variant analysis followed the American College of Medical Genetics and Genomics (ACMG) guidelines ( 5 ). Additionally, in silico analyses predicted the impact of frameshift indels using the SIFT Indel tool ( 6 ). The potential for nonsense-mediated decay (NMD) induced by the variant was assessed with the ‘masonmd’ R package by Hu, Yau, and Ahmed ( 7 ). For structural analysis and visualization, the PyMOL Molecular Graphics System by DeLano Scientific, San Carlos, CA, USA, was used. Index and carries clinical follow-upOur protocol for monitoring asymptomatic MEN1 variant carriers adheres to the Endocrine Society’s guidelines ( 8 ), incorporating biochemical and radiological tests specified for different tumor types from certain ages, as outlined in Tables 1 and 2 . For parathyroid tumors, annual screenings of calcium and PTH start at age eight, bypassing imaging. DP-NET screenings—gastrinomas from age 20 with gastrin and gastric pH tests, insulinomas from age 5 with fasting glucose and insulin levels, and other NETs from age 10 with Chromogranin-A and additional markers—are supported by yearly imaging (MRI, CT, or EUS). Pituitary tumors are screened from age 5 using prolactin and IGF-I levels, plus triennial MRI scans. Adrenal screenings, initiating at age 10, involve yearly MRI or CT scans, triggered by specific symptoms or when detecting tumors exceeding 1 cm.  Table 1 Clinical and biochemical findings in family members with MEN1 during the follow-up.  Table 2 Biochemical findings in patients with functional DP-NETs. Patients and resultsIndex case iii-2. The index patient, III-2, a 24-year-old female, exhibited symptoms of hyperinsulinemic hypoglycemia for a year before being diagnosed in March 2009. Symptoms included asthenia, adynamia, syncopal episodes, and a seizure a month prior to diagnosis. An endoscopic ultrasound identified a focal hypoechoic lesion with regular edges, measuring 18 mm, located at the pancreas’s head and body junction. In April 2009, a partial pancreatectomy and splenectomy were performed, revealing a multifocal, well-differentiated grade 1 NET according to the WHO 2000 criteria. Reclassification under the WHO 2022 criteria would correspond to a Grade 1 neuroendocrine tumor (well-differentiated, 0 mitoses per 2 mm², and a Ki67 proliferation index of 1%). The immunohistochemistry (IHC) studies performed showed the tumor was positive for CKA1AE3, chromogranin, synaptophysin, and focal positivity for insulin. The mitotic rate was 0 per 2 mm², and the Ki67 index was 1%, indicative of an insulinoma. Examination confirmed no necrosis, vascular invasion, or extrapancreatic extension. The pancreatic section edge, spleen, and a peripancreatic lymph node were all tumor-free. Examination confirmed no necrosis, vascular invasion, or extrapancreatic extension. The pancreatic section edge, spleen, and a peripancreatic lymph node were all tumor-free. After eight months free of hypoglycemic symptoms, the patient developed tonic-clonic seizures and hyperinsulinemic hypoglycemia again. New abdominal imaging showed another lesion in the pancreatic head. An octreotide scintigraphy (99mTc-OCTREOTIDE HYNIC) did not show any hypercaptating lesions expressing somatostatin receptors in the gastroenteropancreatic region or other organs (no image available). This led to further surgery at the Instituto Nacional de Cancerología, Bogotá D.C., Colombia, including a residual pancreatectomy, duodenal resection, and peripancreatic soft tissue resection in May 2010. Pathology confirmed a well-differentiated, grade 1 NET (WHO 2010 criteria), with two lymph nodes involved, one positive for gastrin and ACTH ( Table 2 ). The subsequent development of PHPT was surgically addressed with subtotal parathyroidectomy and reimplantation. After this intervention, the patient required insulin therapy for the management of post-surgical diabetes mellitus. The identification of multiple DP-NETs and parathyroid adenomas prompted the suspicion of MEN1, resulting in a referral for genetic testing in July 2010. Sanger sequencing identified a novel heterozygous variant in the MEN1 gene: c.698dup, p.Met233IlefsTer4. This finding initiated CGS within the family ( Figure 1A ).  Figure 1 MEN1 variant analysis and menin protein structure in a colombian family. (A) Family pedigree across four generations highlighting the index patient (arrow). (B) Forward Sanger sequencing results for the proband (III-2), the proband’s father (II-4), and the proband’s daughter (IV-1), indicating the presence of the MEN1: c.698dup frameshift variant. (C) Three-dimensional crystal structure of human Menin (PDB ID: 3U84), frontal and back views. Domains are color-coded: N-terminus (light pink), Thumb (light violet), Palm (pale yellow), with regions affected by MEN1: c.698dup, p.Met233IlefsTer4 highlighted in gray. The critical Met233-to-isoleucine mutation within the compromised domains is marked in red, affecting the Palm and complete Finger domain, and illustrating the disruption’s impact on MIPs interaction regions. MIPs, Menin interaction proteins. Created with BioRender.com . The patient lost contact in 2011 due to pregnancy but returned in 2013 with hyperglycemia and severe hypokalemia, leading to a diagnosis of ACTH-dependent ectopic Cushing’s syndrome (EAS). Imaging (abdominal and pelvic MRI) showed focal hepatic lesions and pelvic bone involvement but no pancreatic or thoracic lesions. An octreotide scintigraphy (99mTc-OCTREOTIDE HYNIC) performed in June 2013 before the surgical intervention did not show any lesions expressing somatostatin receptors. Treatment with ketoconazole partially controlled hypercortisolism, and a left hemihepatectomy with periportal lymph node resection was performed in June 2013 to address liver compromise. Pathology revealed liver and periportal lymph node metastases from a poorly differentiated small cell neuroendocrine carcinoma (WHO 2010 criteria). Reclassification under the WHO 2022 criteria would correspond to a small cell neuroendocrine carcinoma (25 mitoses per 2 mm² and a Ki67 index of 50%), with focal ACTH positivity in both the liver lesion and metastatic nodes. Other causes of ACTH-producing tumors were excluded by performing comprehensive imaging studies, including brain MRI, chest CT, and abdominal-pelvic MRI. Additionally, an octreotide scintigraphy (HYNIC TOC) was conducted, revealing no other tumor lesions apart from those in the liver and a bone lesion in the pelvis. A PET 68-Ga-Dota scan was not performed due to unavailability, but a HYNIC TOC scintigraphy was done prior to the left hemi-hepatectomy, and the liver lesion did not express somatostatin receptors, confirming the cause of EAS as ACTH-producing tumors. Despite external radiotherapy and chemotherapy with etoposide/cisplatin, the disease progressed to involve the liver, bones, and lungs, leading to the patient’s death five years post-diagnosis. Variant analysis and curationThe novel 698-base pair (T) duplication in the MEN1 gene was identified via Sanger sequencing ( Figure 1B ), resulting in a significant frameshift variant designated as MEN1 (NM_000244): c.698dup, p.Met233IlefsTer4 ( Figure 1B ). This frameshift variant affects transcript NM_000244.3 (NCBI) and an alternative transcript, ENST00000377316.6 (Ensembl). It remains unreported in ClinVar, the French MEN1 database ( http://www.umd.be/MEN1/ ), and other genomic variation databases (1000Genomes, ExAC, dbSNP, and HGMD). Protein modeling, as shown in Figure 1C , predicted a compromise in the Palm and Finger domain of the Menin protein (PDB ID: 3U84), which affects the Pocket region—a well-known interaction area with other significant oncogenic proteins. Given the variant’s class (insertion/duplication), we employed the SIFT Indel tool ( 6 ). Using sequence data based on GRCh37/hg19, we predicted that our variant is likely to be damaging, with a confidence score of 0.858. Additionally, this tool uses a basic criterion for predicting nonsense-mediated decay (NMD), suggesting that our variant could trigger NMD due to its location within the transcript. Further analysis with the algorithm developed by Hu, Yau, and Ahmed ( 7 ), using ENTREZ ID: 4221, classified the variant as likely to elicit NMD. This classification takes into account criteria such as having more than two exons (our case 9 exons), the distance of the stop codon from the last exon-exon junction being greater than 50 bp (in our case, 1365 bp), and the premature termination codon (PTC) being located more than 200 bp downstream from the start codon (in our case, 856 bp), resulting in a mutated coding sequence length of 1850 nt. Our variant meets these criteria, further supporting its potential to initiate NMD. Following the analysis, the variant was initially classified as “likely pathogenic” based on the phenotype of the index case, aligning with PVS1 and PM2 criteria. Subsequently, the detailed follow-up, which demonstrated the variant’s familial segregation (meeting the PP1 criterion) and the appearance of MEN1 symptoms in carriers (fulfilling the PP4 criterion), led to an upgrade in its classification to “pathogenic.” This revision highlights the significant correlation between the observed clinical features and a disease with a genetic foundation ( 9 ). Cascade genetic screeningFrom 2010 to 2022, a cascade genetic screening, preceded by pretest genetic counseling, was conducted on eight family members spanning three generations ( Figure 1A ), utilizing Sanger sequencing. This screening uncovered four asymptomatic carriers of the pathogenic variant MEN1 : c.698dup, p.Met233IlefsTer4. The initial screening in 2010 focused on the daughters of the index case, leading to the identification of one carrier (IV-2). Further testing of her parents revealed that her mother was not a carrier, while her father (II-4) was found to be a carrier in 2013. In the same year, screenings were extended to the paternal aunts of the index case, all of whom tested negative. However, the fraternal sister of the index case was also identified as a carrier. After II-4’s remarriage and the birth of a son, the child, given the family’s medical history, was tested in 2021 and confirmed as a carrier of the MEN1 variant. Following the identification of this variant, clinical follow-up started after post-test genetic counseling was provided ( Figure 2 ).  Figure 2 Timeline of molecular and clinical manifestations of MEN1 in the colombian family with MEN1: c.698dup. *index case. Created with BioRender.com . Follow-up MEN1 carriersPatient II-4, the father of the index case, was identified as a MEN1 variant carrier on June 21, 2013 ( Figure 2 ), at 48 years old but remained asymptomatic until 2020, at age 55, when he began experiencing dyspepsia and self-limited diarrhea. This led to the discovery of hypergastrinemia. An upper digestive tract endoscopy uncovered a nodular lesion in the duodenum, and a biopsy confirmed well-differentiated, multifocal, grade I NETs. Abdominal MRI later revealed a lesion adjacent to the uncinate process and another near the third portion of the duodenum, both without compressive effects, along with an intramural lesion in the second portion of the duodenum. The disease was considered unresectable not only due to the duodenal mass but also because of conglomerated lymph nodes around the pancreas and duodenum. Given the inoperability, treatment was initiated with proton pump inhibitors and Lanreotide, stabilizing the disease for a year. However, in January 2022, disease progression was observed, leading to the administration of four cycles of lutetium-177 DOTATOC treatment, totaling a dose of 800 mCi by September 2022, which resulted in stable disease six months post-treatment. Additionally, in early 2021, PHPT was diagnosed, leading to a left lower parathyroidectomy in March 2021. Histopathological examination confirmed a parathyroid adenoma, achieving biochemical control post-surgery. As of the last follow-up, there has been no documented pituitary involvement. Patient III-3, the sister of the index case, was identified as a MEN1 variant carrier in June 2013 at 21 years old ( Figure 2 ). Three years after her identification, she was diagnosed with a prolactin-secreting pituitary microadenoma, prompting the initiation of cabergoline treatment. Concurrently, PHPT was diagnosed, leading to the surgical intervention of right lower parathyroidectomy. These treatments successfully controlled her prolactin and calcium levels. In June 2020, a follow-up pituitary MRI revealed no visible lesion, leading to the discontinuation of cabergoline. However, six months later, she experienced a recurrence of symptomatic hyperprolactinemia, necessitating the resumption of cabergoline therapy. Now, at 31 years old, she remains under treatment, and as of the latest assessments, no duodenal-pancreatic neuroendocrine tumors (NETs) have been detected. Patient III-4 was confirmed as a MEN1 variant carrier in March 2022 at 22 ( Figure 2 ). Following the discovery, the patient consulted with endocrinology and genetic services. Eight months after variant identification, screening unveiled a lobulated pituitary adenoma measuring 9 x 9 x 9 mm. Biochemical evaluations indicated excesses in growth hormone (GH) and prolactin. Consequently, a transsphenoidal surgical approach was employed to remove the pituitary lesion within the same year. Histology of the excised tissue revealed a pituitary microadenoma with a focal expression of prolactin and growth hormone and a Ki67 proliferation index of 3%. Following surgery, the patient developed central hypocortisolism, necessitating the initiation of hydrocortisone supplementation. Despite surgery, persistently elevated insulin-like growth factor 1 (IGF-1) and GH levels were observed, leading to the prescription of somatostatin analog treatment. In parallel, PHPT was diagnosed with scintigraphy detecting a left lower parathyroid lesion. The patient is currently awaiting surgical intervention for this condition. As of the last evaluation, no duodenal-pancreatic NETs have been identified. Patient IV-1 ( Figure 2 ), the daughter of the index case, was confirmed as a MEN1 variant carrier in December 2010 when she was only one year old. Now, at the age of 14, she continues to be asymptomatic, with no tumors detected through ongoing monitoring. Here, we present clinical monitoring of a Colombian family where a novel frameshift variant in the MEN1 gene is present across three generations. The variant, MEN1: c.698dup, p.Met233IlefsTer4, was classified as pathogenic using ACMG criteria and in silico tests and observed clinical features within the family. Although frameshift variants constitute the most prevalent alterations in MEN1, accounting for 42% of cases ( 10 ), the precise molecular mechanisms underlying their impact remain varied and undefined. Welch et al. ( 11 ) described a frameshift variant, c.674delG, located in exon 4 of MEN1, merely 8 codons proximal to our identified variant. This variant was identified in four family members diagnosed with PHPT and DP-NETs, with one member also presenting additional tumor types. Despite certain clinical resemblances with our case, the manifestations in our family exhibit greater variability, which might be attributed to variances in clinical follow-up protocols and the specific molecular mechanisms of the variant. Welch et al. ( 11 ) attributed the clinical impacts of their variant to a loss of interaction (LOI) with proteins like FANCD2 and FOXN3. This mechanism could similarly apply to our variant (see Figure 1C ), as it compromises the palm domain, affecting the Pocket region—a well-known site associated with interaction with most of the menin interaction proteins (MIPs) involved in the oncogenic process in MEN1 ( 12 ). However, since truncating menin proteins are frequently undetectable in MEN1 patients, frameshift variants like ours are likely subject to nonsense-mediated decay (NMD), positioning NMD as the primary disease mechanism in this context. Supporting this theory, our in silico analyses strongly predict NMD for the c.698dup variant, rather than LOI. This underscores the molecular diversity of frameshift variants in MEN1 and helps elucidate the variability in clinical presentations observed in families affected by MEN1. The family described herein exhibits classical MEN1 phenotypes, including PHPT in 4 out of 5 carriers, and various neoplasms such as DP-NETs in 2 out of 5 carriers, and PitNENs in 2 out of 5 carriers ( 3 ). However, notable variations in phenotype were observed. Regarding DP-NETs, we observed an unusual deviation from the typical non-functional tumor presentation, which usually occurs at a rate of 40% ( 13 ). Notably, we identified two instances of functional NETs within a single family. Specifically, Case II-4 was diagnosed with Zollinger-Ellison syndrome at age 48. Moreover, the index case initially presented with an insulinoma at 24, experienced a relapse at 25, and exhibited metastatic spread to two lymph nodes, with one testing positive for gastrin and ACTH. This case evolved into Cushing’s syndrome due to ectopic ACTH secretion syndrome (EAS) provoked by liver metastasis originating from a pancreatic neuroendocrine carcinoma (NEC), an occurrence not previously documented in MEN1 literature for pancreatic NECs. Ectopic ACTH secretion syndrome (EAS) is typically associated with bronchial, lung, and thymus neuroendocrine neoplasms ( 14 ) and is rare about DP-NETs. Thus, our index case underscores the significance of this clinical manifestation within the context of MEN1, presenting, to our knowledge, the first reported case of MEN1 with this specific clinical presentation. Neuroendocrine carcinomas (NECs), whether small-cell (SCNECs) or large-cell (LCNECs), exhibit aggressive clinical behavior and poor prognosis, characterized by high-grade nuclear features, high mitotic counts (often >20 per 2 mm²), and a Ki-67 index usually exceeding 55% ( 15 ). They are rarely associated with hormonal syndromes. However, the index case features a functional ACTH-producing pancreatic-SCNEC (P-SCNEC), a rarity in MEN1-associated NETs, suggesting potential tumor grade progression from a well-differentiated DP-NET. This observation underscores the dynamic evolution of MEN1-associated tumors and highlights the complexity and heterogeneity of pNETs, including variations in histopathological grade, hormone secretion, and genetic alterations ( 13 , 16 ). The phenomenon of grade progression, more common in metachronous than synchronous metastases, further emphasizes the evolving nature of these tumors ( 17 ). In our index case, this progression could be related to the rapid progression despite external radiotherapy and chemotherapy. On the other hand, Pituitary Neuroendocrine Neoplasms (PitNENs) occur in 30% to 50% of individuals with MEN1 ( 18 ), typically emerging between the fourth and sixth decades ( 19 ). While PitNETs are usually macroadenomas (85%), our findings include two family members, III-3 and III-4, aged 23 and 25 respectively, with microadenomas, mainly as prolactinomas, and in one instance, with additional GH secretion, a rarity in only 5% of PitNETs ( 20 ). This finding of microadenomas, diverging from the typical macroadenoma presentation in MEN1, suggests the benefits of early detection through proactive screening. Early identification allows for more straightforward surgical management and minimizes neurological impacts, emphasizing the advantage of screening before the appearance of clinical symptoms for better management and outcomes. This family report underscores the complexity and dynamic progression of MEN1, unveiling the identification of a functional ACTH-producing P-SCNEC as a novel entity within the spectrum of MEN1-associated DP-NETs. Additionally, we report the discovery of a pathogenic frameshift variant, c.698dup, in the MEN1 gene. This variant is subject to NMD, which may explain the varied clinical manifestations seen in this family. While our findings emphasize the importance of cascade screening in facilitating early diagnosis and individualized treatment of MEN1, it is essential to note that our conclusions are based on a single-family cohort. Therefore, the significance of these findings should be considered with caution and in the context of broader studies. Nonetheless, proactive cascade genetic screening remains crucial for identifying asymptomatic carriers, optimizing patient care, and improving outcomes before the disease progresses to more critical stages. Data availability statementThe original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author. Ethics statementThis study was approved by the Institutional Review Board of the Instituto Nacional de Cancerología. It was conducted in accordance with local legislation and institutional requirements. Written informed consent was obtained from the individuals for the publication of any potentially identifiable images or data included in this article. Author contributionsJR: Conceptualization, Data curation, Formal analysis, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AG: Conceptualization, Formal analysis, Investigation, Supervision, Writing – original draft, Writing – review & editing. WT: Conceptualization, Data curation, Formal analysis, Supervision, Validation, Visualization, Writing – review & editing. VM: Methodology, Validation, Writing – review & editing. AR: Methodology, Validation, Writing – review & editing. IV: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. JA: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. JB: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. JB: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. OT: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. LM: Data curation, Formal analysis, Investigation, Methodology, Supervision, Validation, Writing – review & editing. The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The Universidad Cooperativa de Colombia provided financial support for the publication of this article. Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher. 1. Pardi E, Borsari S, Saponaro F, Bogazzi F, Urbani C, Mariotti S, et al. Mutational and large deletion study of genes implicated in hereditary forms of primary hyperparathyroidism and correlation with clinical features. PloS One . (2017) 12:e0186485. doi: 10.1371/journal.pone.0186485 PubMed Abstract | CrossRef Full Text | Google Scholar 2. Matkar S, Thiel A, Hua X. Menin: a scaffold protein that controls gene expression and cell signaling. Trends Biochem Sci . (2013) 38:394–402. doi: 10.1016/j.tibs.2013.05.005 3. Kamilaris CDC, Stratakis CA. Multiple endocrine neoplasia type 1 (MEN1): an update and the significance of early genetic and clinical diagnosis. Front Endocrinol (Lausanne) . (2019) 10:339. doi: 10.3389/fendo.2019.00339 4. Mele C, Mencarelli M, Caputo M, Mai S, Pagano L, Aimaretti G, et al. Phenotypes associated with MEN1 syndrome: A focus on genotype-phenotype correlations. Front Endocrinol (Lausanne) . (2020) 11:591501. doi: 10.3389/fendo.2020.591501 5. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med . (2015) 17:405–24. doi: 10.1038/gim.2015.30 6. Hu J, Ng PC. Predicting the effects of frameshifting indels. Genome Biol . (2012) 13:R9. doi: 10.1186/gb-2012-13-2-r9 7. Hu Z, Yau C, Ahmed AA. A pan-cancer genome-wide analysis reveals tumour dependencies by induction of nonsense-mediated decay. Nat Commun . (2017) 8:15943. doi: 10.1038/ncomms15943 8. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab . (2012) 97:2990–3011. doi: 10.1210/jc.2012-1230 9. Biesecker LG, Byrne AB, Harrison SM, Pesaran T, Schäffer AA, Shirts BH, et al. ClinGen guidance for use of the PP1/BS4 co-segregation and PP4 phenotype specificity criteria for sequence variant pathogenicity classification. Am J Hum Genet . (2024) 111:24–38. doi: 10.1016/j.ajhg.2023.11.009 10. Hu WM, Zhang Q, Huang LH, Mo ZH, Long XD, Yang YB, et al. Identification of novel variants in MEN1: A study conducted with four multiple endocrine neoplasia type 1 patients. Horm Metab Res . (2020) 52:788–95. doi: 10.1055/a-1147-1375 11. Welsch C, Flügel AK, Rondot S, Schulze E, Sircar I, Nußbaumer J, et al. Distinct clinical phenotypes in a family with a novel truncating MEN1 frameshift mutation. BMC Endocr Disord . (2022) 22:64. doi: 10.1186/s12902-022-00978-9 12. Huang J, Gurung B, Wan B, Matkar S, Veniaminova NA, Wan K, et al. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature . (2012) 482:542–6. doi: 10.1038/nature10806 13. Jensen RT, Norton JA. Treatment of pancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: some clarity but continued controversy. Pancreas . (2017) 46:589–94. doi: 10.1097/MPA.0000000000000825 14. Lopez-Montoya V, Gutierrez-Restrepo J, Grajales JLT, Aristizabal N, Pantoja D, Roman-Gonzalez A, et al. Ectopic cushing syndrome in Colombia. Arch Endocrinol Metab . (2021) 64:687–94. doi: 10.20945/2359-3997000000271 15. Strosberg JR, Coppola D, Klimstra DS, Phan AT, Kulke MH, Wiseman GA, et al. The NANETS consensus guidelines for the diagnosis and management of poorly differentiated (high-grade) extrapulmonary neuroendocrine carcinomas. Pancreas . (2010) 39:799–800. doi: 10.1097/MPA.0b013e3181ebb56f 16. Tang X, Shao Y, Yi X, Newey PJ, Li D, Ding K, et al. Metastatic timing and genetic heterogeneity in the evolution of a pancreatic neuroendocrine tumor. Am J Gastroenterol . (2021) 116:844–7. doi: 10.14309/ajg.0000000000001004 17. Grillo F, Albertelli M, Brisigotti MP, Borra T, Boschetti M, Fiocca R, et al. Grade increases in gastroenteropancreatic neuroendocrine tumor metastases compared to the primary tumor. Neuroendocrinology . (2016) 103:452–9. doi: 10.1159/000439434 18. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol . (2014) 386:2–15. doi: 10.1016/j.mce.2013.08.002 19. Syro LV, Scheithauer BW, Kovacs K, Toledo RA, Londoño FJ, Ortiz LD, et al. Pituitary tumors in patients with MEN1 syndrome. Clinics (Sao Paulo) . (2012) 67 Suppl 1:43–8. doi: 10.6061/clinics/2012(Sup01)09 20. Vergès B, Boureille F, Goudet P, Murat A, Beckers A, Sassolas G, et al. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab . (2002) 87:457–65. doi: 10.1210/jcem.87.2.8145 Keywords: multiple endocrine neoplasia type 1 (MEN1), cascade genetic screening, neuroendocrine tumors (NETs), frameshift variant, neuroendocrine - carcinoma Citation: Riaño-Moreno JC, González-Clavijo AM, Torres J. WC, Medina B. VL, Romero-Rojas AE, Vieda-Celemin I, Avila-Moya JA, Baron-Cardona JA, Bravo-Patiño JP, Torres-Zambrano OS and Maya LFF (2024) Case report: Comprehensive follow-up of a Colombian family carrying a novel MEN1 variant linked to a rare ACTH-producing pancreatic neuroendocrine carcinoma. Front. Endocrinol. 15:1398436. doi: 10.3389/fendo.2024.1398436 Received: 09 March 2024; Accepted: 08 July 2024; Published: 22 July 2024. Reviewed by: Copyright © 2024 Riaño-Moreno, González-Clavijo, Torres J., Medina B., Romero-Rojas, Vieda-Celemin, Avila-Moya, Baron-Cardona, Bravo-Patiño, Torres-Zambrano and Maya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY) . The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms. *Correspondence: Julián C. Riaño-Moreno, [email protected] † These authors have contributed equally to this work Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher. Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
Hemodynamic responses to the cold pressor test in individuals with metabolic syndrome: a case-control study in a multiracial sample of adults
Journal of Human Hypertension ( 2024 ) Cite this article Metrics details
Previous research shows that exercise pressor and metaboreflex responses are significantly exaggerated in individuals with metabolic syndrome, but it is unclear if these exaggerated responses extend to the cold pressor test (CPT). This study tested the hypothesis that, contrary to previously reported exaggerated responses during exercise, CPT responses would not be significantly exaggerated in individuals with MetS compared to matched controls. Eleven individuals with MetS and eleven control participants matched by age, race, sex, and ethnicity completed a cardiometabolic prescreening and a CPT. Each CPT required participants to immerse their hand in ice water for two minutes while beat-by-beat blood pressure, heart rate (HR), and leg blood flow (LBF) were continuously measured. Leg vascular conductance (LVC) was calculated as LBF divided by mean arterial pressure (MAP). The precent changes in MAP, systolic blood pressure (SBP), diastolic blood pressure (DBP), HR, LBF, and LVC were compared across time (BL vs. Minutes 1 and 2 of CPT) and between groups (MetS vs. Control) using repeated measures analyses of variance. As expected, MAP ( f = 32.11, p < 0.001), SBP ( f = 23.18, p < 0.001), DBP ( f = 40.39, p < 0.001), and HR ( f = 31.81, p < 0.001) increased during the CPT, and LBF ( f = 4.75, p = 0.014) and LVC ( f = 13.88, p < 0.001) decreased. However, no significant main effects of group or group by time interactions were observed ( f ≤ 0.391, p ≥ 0.539). These findings indicate that the hemodynamic responses to the CPT are not significantly exaggerated in MetS, and therefore, previous reports of exaggerated exercise pressor and metaboreflex responses in MetS cannot be attributed to generalized sympathetic overexcitability. This is a preview of subscription content, access via your institution Access optionsSubscribe to this journal Receive 12 digital issues and online access to articles 111,21 € per year only 9,27 € per issue Buy this article
Prices may be subject to local taxes which are calculated during checkout  Similar content being viewed by others Reliability of blood pressure responses used to define an exaggerated blood pressure response to exercise in young healthy adults Lower Systolic Blood Pressure in Normotensive Subjects is Related to Better Autonomic Recovery Following Exercise Effects of aerobic, resistance and concurrent exercise on pulse wave reflection and autonomic modulation in men with elevated blood pressureData availability. The raw data used in this study will be made available by authors upon reasonable request without undue delay and within the regulations of the University of Southern Mississippi. Victor RG, Leimbach WN Jr, Seals DR, Wallin BG, Mark AL. Effects of the cold pressor test on muscle sympathetic nerve activity in humans. Hypertension. 1987;9:429–36. Article CAS PubMed Google Scholar Yamamoto K, Iwase S, Mano T. Responses of muscle sympathetic nerve activity and cardiac output to the cold pressor test. Jpn J Physiol. 1992;42:239–52. Kasagi F, Akahoshi M, Shimaoka K. Relation between cold pressor test and development of hypertension based on 28-year follow-up. Hypertension. 1995;25:71–76. Han Y, Du J, Wang J, Liu B, Yan YL, Deng SB, et al. Cold Pressor Test in Primary Hypertension: A Cross-Sectional Study. Front Cardiovasc Med. 2022;9:860322. Article CAS PubMed PubMed Central Google Scholar Wood DL, Sheps SG, Elveback LR, Schirger A. Cold pressor test as a predictor of hypertension. Hypertension. 1984;6:301–6. Dutra-Marques AC, Rodrigues S, Cepeda FX, Toschi-Dias E, Rondon E, Carvalho JC, et al. Exaggerated Exercise Blood Pressure as a Marker of Baroreflex Dysfunction in Normotensive Metabolic Syndrome Patients. Front Neurosci. 2021;15:680195. Article PubMed PubMed Central Google Scholar Stavres J, Aultman RA, Brandner CF, Newsome TQA, Vallecillo-Bustos A, Wise HL et al. Hemodynamic responses to handgrip and metaboreflex activation are exaggerated in individuals with metabolic syndrome independent of resting blood pressure, waist circumference, and fasting blood glucose. Front Physiol. 2023;14:1212775. Eldridge FL, Millhorn DE, Kiley JP, Waldrop TG. Stimulation by central command of locomotion, respiration and circulation during exercise. Respir Physiol. 1985;59:313–37. Gandevia SC, Macefield VG, Bigland-Ritchie B, Gorman RB, Burke D. Motoneuronal output and gradation of effort in attempts to contract acutely paralysed leg muscles in man. J Physiol. 1993;471:411–27. Amann M, Blain GM, Proctor LT, Sebranek JJ, Pegelow DF, Dempsey JA. Group III and IV muscle afferents contribute to ventilatory and cardiovascular response to rhythmic exercise in humans. J Appl Physiol. 2010;109:966–76. Kaufman MP, Longhurst JC, Rybicki KJ, Wallach JH, Mitchell JH. Effects of static muscular contraction on impulse activity of group III and IV afferents in cats. J Appl Physiol. 1983;55:105–12. Tschakovsky ME, Hughson RL. Rapid blunting of sympathetic vasoconstriction in the human forearm at the onset of exercise. J Appl Physiol. 2003;94:1785–92. Kuniyoshi FH, Trombetta IC, Batalha LT, Rondon MU, Laterza MC, Gowdak MM, et al. Abnormal neurovascular control during sympathoexcitation in obesity. Obes Res. 2003;11:1411–9. Article PubMed Google Scholar Park J, Middlekauff HR, Campese VM. Abnormal sympathetic reactivity to the cold pressor test in overweight humans. Am J Hypertens. 2012;25:1236–41. PubMed PubMed Central Google Scholar Lee BS, Kim KA, Kim JK, Nho H. Augmented Hemodynamic Responses in Obese Young Men during Dynamic Exercise: Role of the Muscle Metaboreflex. Int J Environ Res Public Health. 2020;17:7321. Sayinalp S, Sözen T, Ozdoğan M. Cold pressor test in diabetic autonomic neuropathy. Diabetes Res Clin Pract. 1994;26:21–8. Holwerda SW, Restaino RM, Manrique C, Lastra G, Fisher JP, Fadel PJ. Augmented pressor and sympathetic responses to skeletal muscle metaboreflex activation in type 2 diabetes patients. Am J Physiol Heart Circ Physiol. 2016;310:H300–9. Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, et al. Diagnosis and Management of the Metabolic Syndrome. Circulation. 2005;112:e285–e290. Google Scholar Stavres J, Aultman RS, Brandner CF, Newsome TQA, Vallecillo-Bustos A, Graybeal AJ. Fat-free mass is associated with exercise pressor responses, but not cold pressor responses, in humans: influence of maximal voluntary contraction. Front Sports Active Living. 2024;6:1352192. Faul F, Erdfelder E, Lang AG, Buchner AG. Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175–91. Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2nd edn. New York: Routledge; 1988. Seals DR, Enoka RM. Sympathetic activation is associated with increases in EMG during fatiguing exercise. J Appl Physiol. 1989;66:88–95. Coutsos M, Sala-Mercado JA, Ichinose M, Li Z, Dawe EJ, O’Leary DS. Muscle metaboreflex-induced coronary vasoconstriction functionally limits increases in ventricular contractility. J Appl Physiol. 2010;109:271–8. Kaur J, Machado TM, Alvarez A, Krishnan AC, Hanna HW, Altamimi YH, et al. Muscle metaboreflex activation during dynamic exercise vasoconstricts ischemic active skeletal muscle. Am J Physiol Heart Circ Physiol. 2015;309:H2145–51. Monahan KD, Feehan RP, Sinoway LI, Gao Z. Contribution of sympathetic activation to coronary vasodilatation during the cold pressor test in healthy men: effect of ageing. J Physiol. 2013;591:2937–47. O’Leary DS, Sala-Mercado JA, Hammond RL, Ansorge EJ, Kim JK, Rodriguez J, et al. Muscle metaboreflex-induced increases in cardiac sympathetic activity vasoconstrict the coronary vasculature. J Appl Physiol. 2007;103:190–4. Kalfon R, Campbell J, Alvarez-Alvarado S, Figueroa A. Aortic Hemodynamics and Arterial Stiffness Responses to Muscle Metaboreflex Activation With Concurrent Cold Pressor Test. Am J Hypertens. 2015;28:1332–8. Estrada JA, Ducrocq GP, Kaufman MP. The magnitude of the exercise pressor reflex is influenced by the active skeletal muscle mass in the decerebrate rat. Am J Physiol Regul Integr Comp Physiol. 2020;318:R30–r37. Lee JB, Katerberg C, Bommarito J, Power GA, Millar PJ. Blood Pressure Responses to Post-Exercise Circulatory Occlusion Are Attenuated Following Exercise-Induced Muscle Weakness. Med Sci Sports Exerc. 2023;55:1660–71. Kawakami R, Tanisawa K, Ito T, Usui C, Miyachi M, Torii S, et al. Fat-Free Mass Index as a Surrogate Marker of Appendicular Skeletal Muscle Mass Index for Low Muscle Mass Screening in Sarcopenia. J Am Med Directors Assoc. 2022;23:1955–1961.e3. Article Google Scholar ten Hoor GA, Plasqui G, Schols AMWJ, Kok G. A Benefit of Being Heavier Is Being Strong: a Cross-Sectional Study in Young Adults. Sports Med. 2018;4:12. Hines EA, Brown GE. The cold pressor test for measuring the reactibility of the blood pressure: Data concerning 571 normal and hypertensive subjects. Am Heart J. 1936;11:1–9. Carvalho LP, Di Thommazo-Luporini L, Mendes RG, Cabiddu R, Ricci PA, Basso-Vanelli RP, et al. Metabolic syndrome impact on cardiac autonomic modulation and exercise capacity in obese adults. Auton Neurosci. 2018;213:43–50. Endukuru CK, Gaur GS, Yerrabelli D, Sahoo J, Vairappan B. Impaired baroreflex sensitivity and cardiac autonomic functions are associated with cardiovascular disease risk factors among patients with metabolic syndrome in a tertiary care teaching hospital of South-India. Diabetes Metab Syndr. 2020;14:2043–51. Grassi G, Dell’Oro R, Quarti-Trevano F, Scopelliti F, Seravalle G, Paleari F, et al. Neuroadrenergic and reflex abnormalities in patients with metabolic syndrome. Diabetologia. 2005;48:1359–65. Download references AcknowledgementsThe authors would like to thank Kieron Cox, Diavion Stanfield, Alex Henderson, Havens L. Wise, Brandy Lowe, and Anne Speed for their technical support. This study was supported by the Mississippi Center for Clinical and Translational Research, the National Institutes of Health, and the National Institute of General Medical Sciences (U54GM115428), as well as the University of Southern Mississippi (to J. Stavres and A.J. Graybeal). Author informationAuthors and affiliations. School of Kinesiology and Nutrition, The University of Southern Mississippi, Hattiesburg, MS, USA Jon Stavres, Anabelle Vallecillo-Bustos, Ta’Quoris A. Newsome, Ryan S. Aultman & Austin J. Graybeal Department of Kinesiology, Iowa State University, Ames, IA, USA Caleb F. Brandner You can also search for this author in PubMed Google Scholar ContributionsConceived and designed research: JS and AJG; Performed experiments: JS, RA, TN, AVB, CFB, AJG; Analyzed data: JS, RA, TN, AVB, CFB, AJG; Interpreted results of experiments: JS, RA, TN, AVB, CFB, AJG; Prepared figures: JS; Drafted manuscript: JS; Edited and revised manuscript: JS, RA, TN, AVB, CFB, AJG; Approved final version of manuscript: JS, RA, TN, AVB, CFB, AJG. Corresponding authorCorrespondence to Jon Stavres . Ethics declarationsCompeting interests. The authors declare no competing interests. Ethics approvalThe protocols used in this study were approved by the University of Southern Mississippi internal review board (IRB# 22-1012) and all subjects provided written informed consent. Additional informationPublisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Supplementary informationSupplemental table 1, rights and permissions. Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law. Reprints and permissions About this articleCite this article. Stavres, J., Vallecillo-Bustos, A., Newsome, T.A. et al. Hemodynamic responses to the cold pressor test in individuals with metabolic syndrome: a case-control study in a multiracial sample of adults. J Hum Hypertens (2024). https://doi.org/10.1038/s41371-024-00938-x Download citation Received : 17 April 2024 Revised : 07 July 2024 Accepted : 10 July 2024 Published : 17 July 2024 DOI : https://doi.org/10.1038/s41371-024-00938-x Share this articleAnyone you share the following link with will be able to read this content: Sorry, a shareable link is not currently available for this article. Provided by the Springer Nature SharedIt content-sharing initiative Quick links

Reducing the Gap in Knowledge and Expectations between Clinicians and People with Polycystic Ovary Syndrome or Adrenal Conditions: Simulation via Instant Messaging—Birmingham Advance: Patient and Public Involvement (SIMBA-PPI) Study
BMC Medical Education volume 24 , Article number: 784 ( 2024 ) Cite this article Metrics details To evaluate the efficacy of SIMBA as an educational intervention for both HCPs and people with either PCOS or adrenal conditions and to study the change in knowledge of people with PCOS or adrenal conditions about the conditions and expectations from the HCPs involved in their care following SIMBA-PPI sessions. Two SIMBA-PPI sessions (SIMBA-PPI Polycystic ovary syndrome (SIMBA-PCOS) and SIMBA-PPI Adrenal conditions (SIMBA-Adrenal conditions)) were conducted in September 2021 and March 2022. In both sessions, HCPs interacted with moderators on patient management through WhatsApp. Patients with respective conditions underwent workshop-style learning in the same cases. SIMBA-PCOS transcripts were also translated into Brazilian Portuguese and workshops were held in both Brazilian Portuguese and English. The two groups (HCPs and patients) were then brought together to discuss exploring gaps in knowledge and expectations. The Wilcoxon Signed-Rank test compared differences in pre- and post-SIMBA self-reported confidence levels in HCPs and patients. Qualitative data from the online recordings were transcribed and analysed with inductive thematic analysis to identify gaps in knowledge and expectations from managing the cases. 48 HCPs and 25 patients participated in our study. When compared to pre-SIMBA confidence levels, SIMBA-PPI sessions effectively improved clinicians’ confidence in managing PCOS (40.5%, p < .001) and adrenal conditions (23.0%, p < .001) post-SIMBA. Patient participants’ confidence in HCPs significantly increased in the PCOS session (SIMBA-PCOS: 6.25%, p = 0.01). ConclusionsIntegration of PPI into SIMBA improved HCPs' confidence in managing PCOS and adrenal conditions. SIMBA-PPI also improved patients’ confidence in HCPs. Our findings suggest that participating in SIMBA-PPI sessions can reduce the gap in knowledge and expectations between patients and HCPs involved in their care. Peer Review reports IntroductionPatient and public involvement (PPI) are essential for an equal partnership in clinical decisions and patient-centred care [ 1 ]. The General Medical Council, the regulatory body of doctors in the United Kingdom, first highlighted the importance of PPI in the standards for undergraduate education in “Tomorrow’s Doctors”, published in 2009 and later in the postgraduate ‘Standard for Deaneries’ calling for deaneries to ensure active and meaningful involvement of patients and the public in doctors’ training [ 2 , 3 ]. Until then, patients’ role in education was indirect or passive, with limited contribution to curriculum development and its delivery and student assessment [ 4 , 5 , 6 ]. Yet, amidst this recognition, robust models that effectively integrate established educational theories into PPI initiatives within healthcare settings are lacking. There is a paucity of such activities in doctors’ postgraduate medical education. There is a lack of ontological and epistemological aspects and evidence of reproducibility, scalability, and sustainability in the published PPI models in medical education [ 7 , 8 , 9 ]. Despite the richness of educational theories such as Experiential Learning and Participatory Action Research [ 10 ], their application in developing comprehensive frameworks for PPI within medical education is limited (a brief explanation of all educational theories and frameworks referenced is supplied in Supplementary 1). While theories like the Health Belief Model [ 11 ] and Theory of Planned Behaviour [ 12 ] offer valuable insights into patient behaviours and attitudes, their integration into educational models specifically tailored for fostering patient-provider collaboration is sparse. The potential of the Transtheoretical Model to cater to diverse patient readiness levels for engagement in healthcare decisions remains underexplored. Similarly, empowerment theory [ 13 ] and social cognitive theory [ 14 ], which hold promise in cultivating empowered, informed patient partnerships, have yet to find their place in structured curricula to train future healthcare professionals. This scarcity of models utilising these theories in the context of PPI represents a critical gap in healthcare education. The dearth of comprehensive frameworks that integrate these well-established educational theories stifles the development of healthcare professionals capable of effectively collaborating with, learning from, and empowering patients and communities. Simulation via Instant Messaging—Birmingham Advance (SIMBA) is a virtual simulation platform that successfully improved participating clinicians’ confidence in managing various simulated cases with a reasonable acceptance rate and reproducibility [ 15 , 16 , 17 , 18 ]. SIMBA sessions have been limited to healthcare professionals (HCPs) who underwent simulation-based learning through text messages in WhatsApp, followed by interactive discussions on Zoom. We recently conducted two educational sessions through SIMBA involving HCPs and people affected by PCOS or adrenal conditions. We called these sessions Simulation via Instant Messaging-Birmingham Advance Public and Patient Involvement (SIMBA-PPI). The two SIMBA-PPI sessions were SIMBA-PPI Polycystic ovary syndrome (SIMBA-PCOS) and SIMBA-PPI Adrenal conditions (SIMBA-Adrenal conditions). The SIMBA-PPI model is based on the simulation game and Kolb’s experiential learning theory [ 19 ]. A simulation game is characterised by the outcomes of choices enacted by participants, interconnected within a framework of rules and resource references mirroring real-life scenarios [ 19 ]. David Kolb’s experiential learning theory is a model of human learning with 4 distinct stages [ 20 ]: Stage 1—concrete experience—is enacted by the simulation session whereby HCPs partake in case scenarios, and patients with PCOS and adrenal pathologies undergo the same cases in workshop-style learning Stage 2—reflective observation—is depicted by the post-simulation discussion with experts via Zoom. The HCPs and patients joined the same discussion, allowing a dialogue regarding bridging the knowledge gap between the two groups. Stage 3—abstract conceptualisation—was facilitated by the use of post-SIMBA surveys, which were distributed to all HCPs and patients partaking in SIMBA. Stage 4—active experimenting—is when HCPs and patients can apply their knowledge from the SIMBA simulation sessions to their own lives [ 20 ]. The aims of this study were: To evaluate the efficacy of SIMBA as an educational intervention for both HCPs and people with either PCOS or adrenal conditions To study the change in knowledge of people with PCOS or adrenal conditions about the conditions and expectations from the HCPs involved in their care following SIMBA-PPI sessions. The SIMBA-PCOS session was conducted on 21st September 2021, and the SIMBA-Adrenal conditions session was conducted on 8th March 2022. Both sessions were carried out based on integrating the six-step conceptual framework described by Kern et. al [ 21 ] and the Plan, Do, Study, Act (PDSA) cycle framework (Supplementary 2). This study has received ethics approval from the Science, Technology, Engineering and Mathematics Ethics Committee at the University of Birmingham (ERN_2023-0495). HCPs were recruited via advertisements on the SIMBA website and social media platforms (Facebook and Twitter). PPI participants were recruited through various support groups. Participation was entirely voluntary. Informed consent was taken from each participant. SIMBA-PCOS six-step conceptual framework and PDSA cycle 1Problem identification and general needs assessment. PCOS commonly affects 10–20% of women worldwide [ 22 ]. Following the diagnosis, people with PCOS are often left with unmet information needs, which can further impact their mental health [ 23 , 24 ]. This is probably due to the diverse symptoms of PCOS and the lack of awareness among HCPs and patients regarding the international consensus guidelines for managing people with PCOS [ 25 ]. Current educational interventions towards improving knowledge, attitudes, and practices of PCOS amongst HCPs have limited patient and public involvement. Further, there is a deeply rooted mistrust of HCPs among people with PCOS [ 24 , 26 ]. The collaboration between patients and HCPs can effectively bridge the existing knowledge gap between these two groups. This collaborative approach may encourage greater patient engagement in their healthcare journey and potentially enhance educational initiatives to improve knowledge, attitudes, and practices related to PCOS among healthcare providers. Targeted needs assessmentWe recognised the need for a model to increase understanding of their condition among people with PCOS. Our objective centred on reducing the knowledge gap between HCPs and patients, primarily due to the varied nature of PCOS symptoms, allowing HCPs and affected patients to engage in a dialogue. Goals and objectivesPromote patient and public involvement in clinical decisions and care to achieve an equal partnership between healthcare professionals and patients, Involve people with PCOS in educational interventions to represent and support others, Integrate PPI into medical education at both undergraduate and postgraduate levels. This involves active and meaningful participation of patients and the public in doctors' training. Education strategiesThe SIMBA-PPI model is based on the simulation game and Kolb’s experiential learning theory [ 19 ]. Implementation of the educational model and PDSA Cycle 1Planning cycle 1. This session was divided into two arms: one focused on HCPs with 29 participants and another component involving people with PCOS with 15 participants. The cases for SIMBA PCOS were designed based on PCOS clinical guidelines and a study on the learning needs of people with PCOS and HCPs [ 25 , 27 , 28 ]. These scenarios were converted into anonymised transcripts, with experts' inputs, which included information on past medical history, clinical examinations, laboratory investigations, and imaging results (an example transcript is provided for SIMBA PCOS and SIMBA Adrenal conditions as supplementary 4A-B). Doing cycle 1Moderators for the simulation were recruited through advertisements and trained in each simulated scenario. HCPs interacted with moderators on WhatsApp, taking relevant history and requesting appropriate investigations to make diagnoses and management/follow-up plans. The moderators provided the information requested by the HCPs from the pre-prepared transcripts. If the requested information was unavailable in transcripts, the moderator replied, 'Information requested is not available’. After completing all the simulated cases, participants attended a discussion about the cases with appropriate experts. We previously published a detailed description of the various steps involved in the SIMBA session, and a summary is included in Supplementary 5 [ 18 ]. In parallel, people with PCOS were invited to participate in workshop-style learning. Here, the transcripts of cases used in SIMBA PPI-HCP were presented to participants, sharing information on how people with PCOS were managed in real life. This was followed with a discussion of the instances concentrating on ‘what was done well’, ‘what could be done better’, and ‘what was not covered’ during the case management. A participant was invited to volunteer and summarise each case, which was presented to the other participants during the discussion. Case discussion with Q&AThis session brought together HCPs and people with PCOS to discuss the cases in detail. Thirty minutes were allocated per case. In the first 5 min, experts presented the case to the group. This was followed by a discussion of the cases about evidence-based guidelines (5 min). In the following five minutes, HCPs who participated in the simulation asked their queries on the cases. The participant from the SIMBA PPI-PCOS group who summarised the discussions shared their findings on that case for five minutes. Each session was concluded with a 10-min discussion on the challenges of managing the cases to identify the gap in knowledge and expectations between people with PCOS and their HCPs. In total the case discussion lasted 2.5 h. Evaluation and feedbackStudying cycle 1 and acting cycle 1. We invited feedback from the SIMBA team and the expert chairs of the session. This was combined with the feedback from HCPs and PPI participants. The summary of the feedback is as follows which was implemented: SIMBA team members suggested a serial instead of a parallel approach for HCP and PPI sessions to better distribute the workload. Chairs recommended a pre-session meeting instead of just emails to understand the session better. The main feedback from HCP participants was to conduct a session on various cases. PPI participants recommended either breaking down the cases over multiple sessions or reducing the number of cases to allow for more in-depth exploration enabling equal participation from all attendees and ensure representation from a broader range of patient groups. SIMBA-Adrenal conditions six-step conceptual framework and PDSA cycle 2In contrast, compared to PCOS, most adrenal conditions are classified as rare diseases due to their low prevalence [ 29 , 30 ]. However, if they remain unrecognised, patients can develop adrenal crisis, which can be fatal. Therefore, HCPs must establish effective educational interventions to recognise adrenal conditions early on. Involving those with adrenal conditions can help HCPs improve their communication skills in the diagnosis and management of adrenal conditions. In this session, we considered all the suggestions from the SIMBA-PCOS session and worked to optimise the model to address better our aim of bridging the knowledge gap between patients and HCPs. We identified the need for a model to accommodate better the HCPs and patients participating in the SIMBA PPI study. Instead of both HCP and PPI arms in parallel, people with adrenal conditions were invited on a separate day beforehand. This was implemented following feedback from the SIMBA team members, as detailed above. It also provided more options for PPI members, enabling them to arrange the session at a time of their convenience instead of a fixed slot. This also allowed the SIMBA team more time to summarise the discussions of each case beforehand. Furthermore, we created an opportunity for input from Brazilian Portuguese-speaking communities. To facilitate this, the six case transcripts were translated into Brazilian Portuguese. A SIMBA team member fluent in English and Brazilian Portuguese held the workshop-style learning in Brazilian Portuguese. They transcribed and summarised the discussion from the workshop into English, which was presented at the discussion. We aimed to develop a model that promoted PPI in clinical decisions and allowed an open dialogue between patients and HCPs, improving the clinical care of people with adrenal conditions. As with SIMBA-PCOS, the SIMBA-Adrenal conditions model is based on the simulation game and Kolb’s experiential learning theory [ 19 ]. Implementation of the educational model and PDSA Cycle 2Planning cycle 2. For the session, the cases included Addison’s disease, adrenal incidentaloma, adrenocortical carcinoma, Cushing’s syndrome, mild autonomous cortical secretion, and primary aldosteronism. Instead of both HCP and PPI arms in parallel, people with adrenal conditions were invited on a particular day beforehand. This was implemented following feedback from the SIMBA team members, as detailed above. The rest of the planning was similar to the first cycle, where PCOS scenarios were simulated. Doing cycle 227 HCPs underwent simulation-based learning focusing on six case scenarios of adrenal conditions as detailed in the SIMBA-PCOS session. PPI workshops were held a week before the SIMBA-Adrenal conditions session, and conducted similar to SIMBA-PCOS session. We introduceds several changes to the case discussion compared to the first cycle. ItWe extendeds the total duration to 3.5 hours, compared to 2.5 hours in the first version, by increasing the time allocated per to ensure each case discussion lasted to 30 minutes. NotablyIn contrast to cycle one, each case discussionit beginsan with a 10-minute pre-recorded expert talk to avoid technical issues and save time, unlike the live 5-minute expert presentation in the first version. Additionally, a SIMBA PPI-Adrenal group member provides a 5-minute summary, and the session concludes with a longer, 15-minute open Q&A, involving all expert panel members, allowing for a more comprehensive discussion. These changes aim to streamline the process and enhance the depth of interaction between healthcare professionals and participants. Studying cycle 2We gathered feedback from SIMBA team members, chairs, experts involved in the discussion, and participants. The primary feedbacks were: SIMBA team members felt the session was much better regarding organisation and delivery than the SIMBA-PPI PCOS session. Chairs recommended a follow-up email following the pre-session meeting to summarise the plans for the session. Participants recommended to continue organising similar sessions but aim for weekdays instead of weekends. Acting cycle 2We gathered all the feedback and analysed the data from both sessions to recommend future sessions, as detailed in the results and discussion sections below. Data collection and statistical analysesTo answer our research questions, we used a mixed quantitative and qualitative research method. The analysis included data from participants who completed both pre- and post-SIMBA surveys. HCP participantsBaseline characteristics, including location and level of training, were recorded. The differences in pre- and post-SIMBA questionnaire responses measured the changes in participants' self-reported confidence levels in managing cases included in the SIMBA-PPI session. The impact of participant’s responses to the six domains of medical education based on the core competencies of the Accreditation Council for Graduate Medical Education (ACGME) was also assessed [ 31 ]. Participants could select as many competencies as they found relevant to the session, including patient care, professionalism, knowledge of patient management, system-based practice, practice-based learning, and communication skills. PPI participantsdemographic information including age, ethnicity, location, and years since diagnosis were recorded. The efficacy of SIMBA in increasing knowledge of the respective conditions was explored through a series of questions and measured using a 5-point Likert scale. Pre- and post-SIMBA questionnaires were used to evaluate the efficacy (Supplementary 6A-D). These consisted of questions on knowledge regarding the conditions pre- and post-simulation. Unfortunately, due to the split between the workshop and the HCP simulation, many PPI participants did not attend the discussion session; hence, post-SIMBA questionnaires were not filled. The workshops and discussion sessions were recorded and transcribed via Zoom. The content of the transcripts was closely examined and analysed thematically by two independent study authors. Responses from the transcribed sessions were first read and familiarised, systematically identifying the text's main points and attaching labels/codes to capture the main ideas. Relevant and recurrent codes were then collated into themes inductively (data-driven themes) and reviewed. Statistical analysis of quantitative data was performed using STATA (STATA/SE 17.0 for Mac). Confidence levels are reported in frequencies and percentages and are presented as bar charts (Fig. 3 ). The Wilcoxon Signed-Rank test was used to compare pre- and post-SIMBA self-reported confidence levels. Statistical significance was accepted at a 95% confidence level. In addition, improvement in participants' confidence levels of simulated cases compared to non-simulated cases, followed by all sessions combined, was included in the comparison and reported as above (Figs. 1 and 2 ).  SIMBA-PCOS A ) Illustration of changes in healthcare professionals’ confidence levels for managing simulated vs non-simulated cases. B Illustration of changes in confidence levels of women with PCOS for their confidence in HCPs to manage simulated vs non-simulated PCOS-related issues. C Illustration of changes in confidence levels of women with PCOS for their confidence in HCPs’ awareness of options available for managing simulated vs non-simulated PCOS-related issues  SIMBA-Adrenal conditions: An illustration of changes in healthcare professionals’ confidence levels for managing simulated vs non-simulated cases Post-SIMBA questionnaires for HCP also included open-ended questions regarding the overall feedback of the session. We performed a thematic inductive analysis for these open-ended questions to identify common feedback themes. Patient and public involvement statementVarious patient support groups and public were involved since the start of the project. Patients and public underwent workshop-style learning in parallel, using the same cases. HCPs, patients and public then came together to discuss the issues with the experts via Zoom. This promoted patient and public involvement in clinical decisions and care to achieve an equal partnership between healthcare professionals and patients alongside integrating PPI into medical education at both undergraduate and postgraduate levels. This model was codesigned by a PPI expert coordinator (CG). HCP ResultsTwenty-five out of 29 participants (86%) completed both pre- and post-SIMBA questionnaires. Characteristics of participants are described in Supplementary 7. Clinicians’ confidence in managing PCOS-related issues increased post-SIMBA for all eight domains of PCOS measured in this session (skin, weight, fertility, menstruation, menopause, diabetes, mental health, and endometrial cancer). Simulated cases and non-simulated cases showed a 41.0% ( p < 0.001) and 40.0% ( p < 0.001) increase in confidence, respectively, with all cases combined showing a 40.5% increase in confidence ( p < 0.001) (Fig. 1 A). There was an improvement in patient care (64.0%), communication skills (40.0%), professionalism (28.0%), knowledge of patient management (72.0%), system-based practice (32.0%), and practice-based learning (60.0%) (Fig. 2 ). 88.0% of SIMBA-PCOS participants strongly agreed/agreed that simulated topics applied to their practice, and 100% would attend similar SIMBA sessions in the future. Content analysis of feedback from SIMBA-PCOS participants to open-ended questions showed a positive rating and constructive comments for future sessions. Suggestions focused on three themes: time considerations, Q&A discussion format, and session content. Participants reflected on requiring more time for cases and discussion to allow for more questions to be answered. There was a call for more focus on assessing and managing the cases during the Q&A discussion. SIMBA-Adrenal conditionsTwenty-three out of 27 (85%) HCP participants completed both pre- and post-SIMBA questionnaires. Characteristics of participants are described in Supplementary 7. Self-reported confidence increased post-SIMBA-Adrenal conditions for all ten adrenal conditions measured in the session (Addison’s disease, adrenal incidentaloma, adrenocortical carcinoma, Cushing’s syndrome, minimal autonomous cortisol secretion, primary aldosteronism, bilateral adrenal hyperplasia, congenital adrenal hyperplasia, phaeochromocytoma, and virilisation). Simulated cases and non-simulated adrenal cases showed a 22.5% ( p < 0.001) and 24.0% ( p < 0.001) increase in confidence, respectively, with all cases showing a 23.0% increase in confidence ( p < 0.001) (Fig. 2 ). Participants reported improvement in confidence in practice-based learning (69.6%), system-based practice (43.5%), knowledge of patient management (87.0%), professionalism (43.5%), communication skills (26.1%), and patient care (43.5%) (Fig. 3 ). 95.6% strongly agreed/agreed that the simulated cases applied to their practice, and 100% would attend similar SIMBA sessions. Content analysis of feedback from SIMBA-Adrenal conditions participants to open-ended questions showed positive and constructive themes. Suggestions for improvement focussed on session length and structure, with participants reflecting on having smaller groups between chairs and participants for Q&A and using fewer cases to allow for a more in-depth discussion.  HCP’s Self-reported increase in confidence across competencies post-SIMBA-PCOS and SIMBA-Adrenal conditions Patient resultsFifteen women with PCOS attended the session. Twelve (80%) completed both pre- and post-session surveys. Supplementary 8 shows the demographics of women with PCOS who participated in the session. The median age of PCOS diagnosis ( n = 10) was 20.5 (IQR 19–25) years. 83.3% ( n = 10/12) of people were aware of the criteria for diagnosing PCOS, and no significant difference after the session was noted ( p = 1.000). People with PCOS reported their confidence in HCPs increased by 6.25% post-session ( p = 0.0141) (Fig. 1 B). Similarly, people with PCOS’ confidence in HCPs’ awareness of options for managing PCOS-related issues increased by 17.7% ( p = 0.0002) (Fig. 1 C). 90% strongly agreed/agreed that SIMBA-PCOS benefit people with PCOS to better understand their condition and clinicians’ perspectives on the diagnosis and management of PCOS. In addition, 100% of people with PCOS strongly agreed/agreed that SIMBA-PCOS benefits clinicians in understanding patients’ perspectives on diagnosing and managing PCOS. Content analysis on open-ended questions was performed on all 12 participants who completed the post-SIMBA survey. Table 1 summarises these findings. Participants found the session informative, with engaging discussion and organised structure. Suggestions for improvement for future sessions centred around the format of the session advising breaking down the cases over sessions or reducing the number of cases to allow for future exploration of cases, together with organising structured feedback for future session discussions, allowing the equal contribution of all participants and representation from a broader range of patient groups. Ten people diagnosed with adrenal conditions attended SIMBA-Adrenal conditions, and all 10 (100%) completed post-SIMBA surveys. Adrenal conditions represented were Addison's disease ( n = 5), adrenocortical carcinoma ( n = 3), Cushing’s disease ( n = 1), and primary aldosteronism ( n = 1). The median age of diagnosis with an adrenal condition was 39 (IQR 33–53) years. After the session, 70% knew the criteria for diagnosing their adrenal condition. 90% strongly agreed/agreed that SIMBA-Adrenal conditions benefited people with various adrenal conditions to understand their conditions better. 80% thought the session helped them understand clinicians’ perspectives on diagnosing and managing relevant adrenal conditions. Additionally, 80% of participants strongly agreed/agreed that SIMBA-Adrenal conditions benefit clinicians in understanding patients’ perspectives on diagnosing and managing adrenal conditions. Content analysis was performed on all 10 participants’ feedback to open-ended questions. People with adrenal conditions revealed positive comments, including well-presented cases, open information sharing and a shared experience between doctor and patient (Table 2 ). Suggestions for improvement included bringing more doctors to discussion sessions having an agenda for discussion sessions alongside acknowledging individual circumstances and levels of understanding. SIMBA-PPI sessions effectively improved clinicians’ confidence in managing PCOS and adrenal conditions. There was a greater improvement in HCP confidence in managing PCOS compared to adrenal conditions. This is likely due to a lower baseline confidence in managing simulated PCOS compared to adrenal conditions. For both SIMBA-PCOS and SIMBA-Adrenal conditions, most HCPs agreed that the simulated topics were applicable to their practice, and all participants indicated they would attend similar SIMBA sessions in the future. We acted upon HCPs’ feedback from the first SIMBA-PPI session, including allocating more time for case discussions and focussing more on assessment and management during the longer Q&A sessions. Feedback from HCPs for SIMBA-Adrenal was favourable for these changes. We will incorporate the suggestions for smaller groups for Q&A sessions and use fewer cases for more in-depth discussion in future sessions. People with PCOS and adrenal conditions agreed that SIMBA-PPI sessions helped them learn more about their needs and understand clinicians’ perspectives on diagnosis and management. Our findings suggest there is a gap in knowledge and expectations between patients and HCPs involved in their care, and this gap can be reduced by attending SIMBA sessions involving both affected people and HCPs. Drawing from theories such as Experiential Learning [ 20 ] and Participatory Action Research [ 10 ], the SIMBA-PPI model places patients and the public at the forefront. By recognising the importance of community engagement and empowerment, Community-Based Participatory Research [ 32 ] (CBPR) principles are woven into the fabric of the SIMBA-PPI model, emphasising collaboration, relevance, and cultural responsiveness. This educational framework aligns with the Health Belief Model [ 11 ] and Theory of Planned Behaviour [ 12 , 13 ], acknowledging the significance of individual beliefs, attitudes, and readiness to engage in healthcare decisions. At its core, the SIMBA model harnesses the power of Empowerment Theory [ 13 ] and Social Cognitive Theory [ 14 ] to cultivate an educational environment that educates future healthcare providers and empowers patients and the public to participate in their care actively. It champions the development of a new generation of healthcare professionals who value partnership, empathetic communication, and collaboration with patients and communities. To our knowledge, this is the first educational activity with PPI to identify the knowledge gap between HCPs and people with PCOS and adrenal conditions. The efficacy of SIMBA as an educational tool in improving clinicians’ confidence in managing simulated cases, with a high acceptance rate, is consistent with our previous SIMBA sessions on diabetes, endocrinology and acute medicine [ 16 , 18 , 33 , 34 ]. In SIMBA, participants completed three stages of Kolb’s experiential learning cycle before going into the fourth stage—active experimenting -, when they can apply the knowledge learnt from SIMBA into their real-life clinical practice. In SIMBA-PPI, the patients provided additional learning to the HCP participants at each of the three stages of the learning cycle. The experience shared by the patients enabled HCPs to reflectively observe and form a better conceptualisation of the content through lived experiences. Several studies have highlighted the gap in knowledge, attitude, and perception of clinical care between HCPs and people with PCOS and adrenal conditions. Delays in diagnoses of PCOS and inadequate information given to people with PCOS upon diagnosis have been reported worldwide [ 23 , 24 , 35 ]. Poor experiences with diagnoses and management of PCOS have been associated with increased anxiety and depression, further negatively impacting self-management [ 36 , 37 , 38 ]. This has led to patients and their carers relying on mostly non-evidence-based information online, potentially containing inaccuracies and contradicting what patients have been advised by the HCPs [ 36 ]. Similarly, patients with adrenal conditions often receive late diagnoses [ 39 , 40 , 41 ]. Previous studies have also shown that patients were unaware of appropriately adjusting steroid doses during acute illness due to suboptimal and inconsistent education [ 40 , 41 ]. SIMBA-PPI helps to address this issue by bringing HCPs and patients together to share real-life experiences and provides a platform where both can teach and learn from each other [ 36 , 39 ]. Such educational events can also help patients understand evidence-based strategies and clinicians' limitations. This may potentially reduce any unrealistic expectations of the patients and the gap in knowledge and expectations between patients and HCPs. The content analysis of feedback from open-ended questions identified several areas for improvement as part of the PDSA cycle. Further feedback from SIMBA-Adrenal conditions suggests future sessions should be more tailored to individual patient circumstances and different levels of understanding. Additional work is needed to determine whether our findings of increased self-reported confidence levels translate to a long-term improvement in clinical performance and patient-reported outcome measures. The integrative SIMBA model envisions a curriculum where students learn from, with, and about patients and the public. Through immersive, experiential learning opportunities, students engage in authentic healthcare scenarios, fostering critical reflection, and applying insights gained from collaborative interactions. It promotes the creation of inclusive spaces where patients, healthcare professionals, educators, and researchers co-create knowledge and solutions, thereby bridging the gap between theory and practice. In essence, the SIMBA patient public involvement medical education model represents a transformative approach, transcending traditional pedagogical boundaries to nurture a healthcare ecosystem rooted in empathy, collaboration, and patient-centred care. While the study has several strengths in the design and outcomes, we acknowledge that some factors may limit our findings' generalisability, one being that the results are from only two sessions. Our PPI participants were invited through various support groups and were interviewed for their interest in participating. This may be considered as a potential selection bias due to this convenience sampling method. However, the interview was necessary to ensure their engagement for the entire duration of our session. Another limitation was that for SIMBA-Adrenal conditions due to the split between the workshop and the HCP simulation, many PPI participants did not attend the discussion session; hence, post-SIMBA questionnaires were not filled. This can be mitigated in the future by reducing the time gap between the workshop and HCP simulation, ensuring clear communication, sending timely reminders and engaging participants actively in the process. In addition, our small sample size may also impact our results and may not be representative of the general patient population. However, applying a mixed-method approach helped consolidate the findings from the study. The study questionnaires were not validated, which could affect their reliability. Follow-up studies based on the principles of Diffusion of Innovation Theory [ 42 ] and Transtheoretical Model [ 43 ] are needed to study long-term impact. Further studies will look into the differences in opinions, experiences, and expectations of HCPs trained in the UK versus abroad. Similarly, we want to incorporate a wider perspective of patients from different backgrounds. Following the successful implementation of SIMBA in undergraduate education, we are looking to introduce PPI. We also want to implement SIMBA-PPI as a continuing professional development activity for specialist trainees. SIMBA-PPI is an effective learning tool for people with PCOS and adrenal conditions and their healthcare professionals to enhance their understanding of the condition. SIMBA is open access and cost-effective. The model also helped identify and reduce gaps in knowledge and expectations between HCPs and patients with PCOS and adrenal conditions. Future large-scale studies are needed to study the long-term impact on clinical practice and patient-reported outcome measures. Availability of data and materialsData available on request to the corresponding author. Karlsson AW, Janssens A. Patient and public involvement and engagement (PPIE) in healthcare education and thesis work: the first step towards PPIE knowledgeable healthcare professionals. BMJ Open. 2023;13: e067588. Article Google Scholar GMC. Patient and public involvement in undergraduate medical education. General Medical Council (2011). Field S. Tomorrow’s doctors. BMJ. 2009;339: b2696. Wykurz G, Kelly D. Developing the role of patients as teachers: literature review. BMJ : British Medical Journal. 2002;325:818. Towle A, et al. Active patient involvement in the education of health professionals. Med Educ. 2010;44:64–74. Dijk SW, Duijzer EJ, Wienold M. Role of active patient involvement in undergraduate medical education: a systematic review. BMJ Open. 2020;10: e037217. Price A, et al. Frequency of reporting on patient and public involvement (PPI) in research studies published in a general medical journal: a descriptive study. BMJ Open. 2018;8: e020452 Lang I, et al. How common is patient and public involvement (PPI)? Cross-sectional analysis of frequency of PPI reporting in health research papers and associations with methods, funding sources and other factors. BMJ Open. 2022;12: e063356. Arumugam A, et al. Patient and public involvement in research: a review of practical resources for young investigators. BMC Rheumatol. 2023;7:2. Baum, F., MacDougall, C. & Smith, D. Participatory action research. J Epidemiol Community Health (1978) 60 , 854 (2006). Jones CL, et al. The Health Belief Model as an explanatory framework in communication research: exploring parallel, serial, and moderated mediation. Health Commun. 2015;30:566–76. Bosnjak M, Ajzen I, Schmidt P. The Theory of Planned Behavior: Selected Recent Advances and Applications. Eur J Psychol. 2020;16:352. Perkins DD, Zimmerman MA. Empowerment theory, research, and application. Am J Community Psychol. 1995;23:569–79. Bandura A. Social cognitive theory: An agentic perspective. Annu Rev Psychol. 2001;52:1–26. Melson, E. et al. Adaptation and use of media in an innovative simulation-based clinician training programme. BMJ Simul Technol Enhanc Learn 0 , bmjstel-2020–000808 (2021). Morgan G, et al. Utility of Simulation via Instant Messaging – Birmingham Advance (SIMBA) in medical education during COVID-19 pandemic. Journal of the Royal College of Physicians of Edinburgh. 2021;51:168–72. Evans N, et al. SIMBA as an alternative and/or an adjunct to pre-medical work experience during the COVID-19 pandemic. Future Healthc J. 2021;8:e142–5. Melson E, et al. Simulation via instant messaging-Birmingham advance (SIMBA) model helped improve clinicians’ confidence to manage cases in diabetes and endocrinology. BMC Med Educ. 2020;20:1–10. Kriz WC. Creating Effective Learning Environments and Learning Organizations through Gaming Simulation Design. 2003. https://doi.org/10.1177/104687810325820134495-511 . David A. Kolb. Experiential Learning: Experience as the Source of Learning and Development. Prentice-Hall https://scholar.google.com/scholar?hl=en&as_sdt=0,5&cluster=17808912641356781759 (1984). Thomas, P. A., Kern, D. E., Hughes, M. T. & Chen, B. Y. Curriculum Development for Medical Education: A Six-Step Approach . Angewandte Chemie International Edition, 6(11), 951–952. (Johns Hopkins University Press, 2015). Joham AE, et al. Polycystic ovary syndrome Lancet Diabetes Endocrinol. 2022;10:668–80. Gibson-Helm M, Teede H, Dunaif A, Dokras A. Delayed Diagnosis and a Lack of Information Associated With Dissatisfaction in Women With Polycystic Ovary Syndrome. J Clin Endocrinol Metab. 2017;102:604–12. Google Scholar Lau, G. M. et al. A systematic review of lived experiences of people with polycystic ovary syndrome highlights the need for holistic care and co-creation of educational resources. Front Endocrinol (Lausanne) 13 , 1064937 (2022). Teede HJ, et al. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Hum Reprod. 2018;33:1602–18. Elhariry, M., Malhotra, K., Solomon, M., Goyal, K. & Kempegowda, P. Top 100 #PCOS influencers: Understanding who, why and how online content for PCOS is influenced. Front Endocrinol (Lausanne) 13 , 1084047 (2022). Gibson-Helm M, Tassone EC, Teede HJ, Dokras A, Garad R. The Needs of Women and Healthcare Providers regarding Polycystic Ovary Syndrome Information, Resources, and Education: A Systematic Search and Narrative Review. Semin Reprod Med. 2018;36:35–41. Lau, G. M. et al. PCOS pearls - gathering perceptions and opinions from lived experiences of people with polycystic ovary syndrome. Endocrine Abstracts 77 , (2021). Ebbehoj A, et al. Epidemiology of Adrenal Tumors - a Population-based Study in Olmsted County. Minnesota Lancet Diabetes Endocrinol. 2020;8:894. Huecker, M. R., Bhutta, B. S. & Dominique, E. Adrenal Insufficiency. Pediatric Surgery Digest: Second Edition 555–557 (2023) https://doi.org/10.1007/978-3-030-80411-4_36 . Staton, L. J., Kraemer, S. M., Patel, S., Talente, G. M. & Estrada, C. A. Peer chart audits: a tool to meet Accreditation Council on Graduate Medical Education (ACGME) competency in practice-based learning and improvement. Implement Sci 2 , (2007). Collins SE, et al. Community-based participatory research (CBPR): Towards equitable involvement of community in psychology research. Am Psychol. 2018;73:884–98. Wallett, L. et al. Developing a simulation-based learning model for acute medical education during COVID-19 pandemic with Simulation via Instant Messaging - Birmingham Advance (SIMBA). BMJ Open Qual 11 , (2022). Davitadze M, et al. SIMBA: using Kolb’s learning theory in simulation-based learning to improve participants’ confidence. BMC Med Educ. 2022;22:1–11. Hillman, S., Dale, J. & Bryce, C. GPPCOS: exploring women’s experience of the management of PCOS in general practice. British Journal of General Practice 69 , bjgp19X702965 (2019). Avery JC, Braunack-Mayer AJ. The information needs of women diagnosed with Polycystic Ovarian Syndrome - Implications for treatment and health outcomes. BMC Womens Health. 2007;7:1–10. Colwell K, Lujan ME, Lawson KL, Pierson RA, Chizen DR. Women’s perceptions of polycystic ovary syndrome following participation in a clinical research study: implications for knowledge, feelings, and daily health practices. J Obstet Gynaecol Can. 2010;32:453–9. Deeks AA, Gibson-Helm ME, Paul E, Teede HJ. Is having polycystic ovary syndrome a predictor of poor psychological function including anxiety and depression? Hum Reprod. 2011;26:1399–407. Bleicken B, Hahner S, Ventz M, Quinkler M. Delayed diagnosis of adrenal insufficiency is common: a cross-sectional study in 216 patients. Am J Med Sci. 2010;339:525–31. Papierska, L. & Rabijewski, M. Delay in diagnosis of adrenal insufficiency is a frequent cause of adrenal crisis. Int J Endocrinol 2013 , (2013). Shepherd LM, Tahrani AA, Inman C, Arlt W, Carrick-Sen DM. Exploration of knowledge and understanding in patients with primary adrenal insufficiency: A mixed methods study. BMC Endocr Disord. 2017;17:1–10. Kaminski, J. Diffusion of Innovation Theory. Canadian Journal of Nursing Informatics https://cjni.net/journal/?p=1444 (2011). Armitage CJ. Is there utility in the transtheoretical model? Br J Health Psychol. 2009;14:195–210. Download references AcknowledgementsWe thank the HCPs and patients who participated in this study. We thank PCOS and adrenal support groups—PCOS Verity, PCOS vitality, DAISy-PCOS, PCOS Club India, Abaddison Brasil, ACC Support UK, Addison’s Disease Self-Help Group, Alex-The Leukodystrophy Charity, The Association for Multiple Endocrine Neoplasia Disorders and Cushing's UK Facebook group—for their support for the study. We thank the University of Birmingham Medical School students who participated as moderators in this study. We also thank the Health Education West Midlands Specialist trainee committee, Institute of Metabolism and Systems Research, University of Birmingham, and Institute of Applied Health Research, University of Birmingham, for supporting this study. We thank Dr Helena Gleeson, Dr Konstantinos Manalopoulus, Dr Justin Chu, Dr Michael O’Reilly, and Professor Wiebke Arlt for their expert inputs during discussion. Disclosure StatementThe authors report no potential conflict of interest. The study did not receive any funding. Author informationEka Melson, Fatema Rezai and Carina Pan are joint first authors. Authors and AffiliationsInstitute of Clinical Sciences, Imperial College London, London, United Kingdom Institute of Metabolism and Systems Research, University of Birmingham, Birmingham, United Kingdom Eka Melson & Caroline Gillett College of Medical and Dental Sciences, University of Birmingham, Birmingham, United Kingdom Fatema Rezai, Carina Pan, Sung Yat Ng, Ella Blendis, Haaziq Sheikh, Harjeet Kaur, Francesca Pang, Shreya Bhatt, Zahra Olateju, Eloise Radcliffe, Prashanthan Balendran, Abby Radcliffe, Gar Mun Lau & Dengyi Zhou University of Lancaster, Lancaster, United Kingdom Tamzin Ogiliev Walsall Manor Hospital, Walsall, United Kingdom Catherine Cooper Queen Charlotte’s and Chelsea Hospital, London, United Kingdom University of Warwick, Conventry, United Kingdom Farah Abdelhameed Jinnah Medical and Dental College, Karachi, Pakistan Dania Shabbir Clinic NeoLab, Tbilisi, Georgia Meri Davitadze Institute of Applied Health Research, University of Birmingham, Birmingham, United Kingdom Meri Davitadze, Kashish Malhotra & Punith Kempegowda Northwick Park Hospital, London North West University Healthcare NHS Trust, London, United Kingdom Dengyi Zhou Rama Medical College Hospital, Hapur, Uttar Pradesh, India Kashish Malhotra Queen Elizabeth Hospital Birmingham, University Hospitals NHS Foundation Trust, Birmingham, Uttar Pradesh, United Kingdom Punith Kempegowda You can also search for this author in PubMed Google Scholar SIMBA and CoMICs teamContributions. All authors have contributed to the manuscript as per ICMJE criteria. EM, FR, and CP contributed equally to the study and are listed as joint first authors. SYN, TO, EB, JS, HK, CC, FA, FP, SB, DS, ZO, ER, PB, AR, GML, MD, DZ, and HG contributed to the study as moderators and interviewed SIMBA-PPI. KM, JC, MOR, WA, and CG contributed as experts to the conditions studied. PK conceptualised and supervised the delivery of all aspects of SIMBA and critically reviewed the manuscript. The final version has been reviewed and approved by all the named authors. The SIMBA team included members who contributed in all study phases and hence are included as collaborating authors. This includes Emily Warmington, Pavithra Sakthivel, Vina Soran, Anisah Ali, Harshin Pallathoor Balakrishnan, Maiar Elhariry, Sangamithra Ravi, Rachel Nirmal, Aditya Swaminathan, Shams Ali Baig, Isabel Allison, Tamzin Ogiliev, Dwi Delson, Soon Chee Yap, Vardhan Venkatesh, Fazna Rahman, Dr Helena Gleeson, Dr Konstantinos Manalopoulus, Dr Justin Chu, Dr Michael O’Reilly, and Professor Wiebke Arlt . Corresponding authorCorrespondence to Punith Kempegowda . Ethics declarationsCompeting interest. The authors declare no competing interests. Additional informationPublisher’s note. Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Supplementary InformationSupplementary material 1., rights and permissions. Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data. Reprints and permissions About this articleCite this article. Melson, E., Rezai, F., Pan, C. et al. Reducing the Gap in Knowledge and Expectations between Clinicians and People with Polycystic Ovary Syndrome or Adrenal Conditions: Simulation via Instant Messaging—Birmingham Advance: Patient and Public Involvement (SIMBA-PPI) Study. BMC Med Educ 24 , 784 (2024). https://doi.org/10.1186/s12909-024-05772-w Download citation Received : 18 March 2024 Accepted : 11 July 2024 Published : 22 July 2024 DOI : https://doi.org/10.1186/s12909-024-05772-w Share this articleAnyone you share the following link with will be able to read this content: Sorry, a shareable link is not currently available for this article. Provided by the Springer Nature SharedIt content-sharing initiative
BMC Medical EducationISSN: 1472-6920
Chromosomal instability in a patient with ring chromosome 14 syndrome: a case report
Molecular Cytogenetics volume 17 , Article number: 17 ( 2024 ) Cite this article 131 Accesses Metrics details Ring chromosome 14 syndrome is a rare disorder primarily marked by early-onset epilepsy, microcephaly, distinctive craniofacial features, hypotonia, intellectual disability, and delay in both development and language acquisition. Case presentationA 21-year-old woman with a history of epileptic seizures since the age of 1.5 years presented with distinctive craniofacial features, including a prominent and narrow forehead, sparse and short eyebrows, palpebral ptosis, horizontal palpebral fissures, a broad nasal bridge, a prominent nasal tip, a flat philtrum, hypertelorism, midfacial hypoplasia, horizontal labial fissures, a thin upper lip, crowded teeth, an ogival palate, retrognathia, and a wide neck. Additional physical abnormalities included kyphosis, lumbar scoliosis, pectus carinatum, cubitus valgus, thenar and hypothenar hypoplasia, bilateral hallux valgus, shortening of the Achilles tendon on the left foot, and hypoplasia of the labia minora. Chromosomal analysis identified a ring 14 chromosome with breakpoints in p11 and q32.33. An aCGH study revealed a ~ 1.7 Mb deletion on chromosome 14qter, encompassing 23 genes. Genomic instability was evidenced by the presence of micronuclei and aneuploidies involving the ring and other chromosomes. The clinical features of our patient closely resembled those observed in other individuals with ring chromosome 14 syndrome. The most important point was that we were able to verify an instability of the r(14) chromosome, mainly involving anaphasic lags and its exclusion from the nucleus in the form of a micronucleus. Human ring chromosomes are structural abnormalities resulting from two breaks in the DNA strand that fuse to form a circular DNA molecule, often leading to genetic material loss. These chromosomes can possess one or more centromeres or lack one entirely, affecting their segregation during anaphase. Ring chromosomes have been reported for all human chromosomes, with approximately half involving acrocentric chromosomes, including chromosome 14 [ 1 ]. Numerous cases of chromosome 14 rings have been documented, with the deleted region’s size ranging from 0.3 to 5 megabases (Mb) [ 2 ]. The severity of the phenotype, known as ring chromosome 14 syndrome [r(14) syndrome, Online Mendelian Inheritance in Man (OMIM) #616,606], varies among patients [ 3 ]. Key clinical features include early-onset epilepsy, microcephaly, hypotonia, intellectual disability, developmental and language delays, and distinctive craniofacial dysmorphisms such as hypertelorism, micrognathia, a thin upper lip, down-turned mouth corners, a high-arched palate, and large, low-set ears [ 4 ]. Additional features include motor skill issues, retinal abnormalities, feeding difficulties, and behavioral disorders [ 2 , 3 , 5 ]. The deletion of specific genes on 14q32.2-32.3 is believed to cause the primary clinical features of the syndrome [ 3 ], although other genes outside the deleted region might also influence the phenotype [ 6 ]. Ring chromosomes exhibit inherent instability due to the sister chromatid exchange process, which can create dicentric or interlocked ring chromosomes. During mitosis, these chromosomes can undergo anaphase lagging, nondisjunction, or fragmentation, leading to cells without the ring chromosome, cells with multiple ring chromosomes, binucleated cells, internuclear bridges, nuclear protrusions, and micronuclei (MN). This phenomenon is known as tissue-specific dynamic mosaicism [ 7 ]. Ring chromosomes show variable instability in vivo based on their size and genetic content [ 8 ], but there is no clear correlation between the size of a ring chromosome, the occurrence of dynamic mosaicism, and clinical severity [ 9 ]. In this report, we present the case of a female with a ring chromosome 14, who exhibited epileptic seizures, craniofacial dysmorphism, skeletal abnormalities, and genital alterations. Chromosomal instability was also observed through the presence of MN and aneuploidies. A 21-year-old woman, the fifth child of healthy, unrelated parents, experienced epileptic seizures beginning at 1.5 years old, which were managed with valproic acid. She has been seizure-free since the age of 2 and has been off treatment since the age of 8. Currently, she has a height of 162 cm (50th percentile) and a head circumference of 53.5 cm (10th-25th percentile), along with a high anterior hairline. She exhibits craniofacial dysmorphism, including a prominent and narrow forehead, sparse and short eyebrows, palpebral ptosis, horizontal palpebral fissures, a broad nasal bridge, a prominent nasal tip, a flat philtrum, hypertelorism, midfacial hypoplasia, horizontal labial fissures, a thin upper lip, crowded teeth, an ogival palate, retrognathia, and a wide neck. Additional features include kyphosis, lumbar scoliosis, bilateral cubitus valgus, thenar and hypothenar hypoplasia, bilateral hallux valgus, shortening of the Achilles tendon on the left foot, and hypoplasia of the labia minora. Although clinical images are not provided due to lack of parental authorization, her clinical characteristics align with those typically reported for ring chromosome 14 syndrome (Table 1 ). Conventional cytogenetic studiesPeripheral blood lymphocytes from the patient were cultured and harvested using standard methods for karyotype analysis. Chromosomal analysis of GTG-banded metaphases revealed the presence of a ring chromosome 14 (Fig. 1 A). Both parents had a normal karyotype. FISH (fluorescense in situ hybridization) analysis using the TCL1 break-apart and IGH/BCL2 probes indicated the breakpoint occurred distal to the TCL1 (q32.13) but proximal to the IGH (q32.33) genes (Fig. 1 B and D). Additionally, FISH with a nucleolus organizer region (NOR) specific probe showed no signal in the r(14) chromosome (Fig. 1 C). The frequencies of cells with TCL1 break-apart signals in interphase FISH were as follows: 198/243 cells (81.5%) had two signals, 30/243 cells (12.3%) had only one signal, suggesting the loss of the r(14) chromosome, and 15/243 cells (6.2%) had three signals, likely indicating a duplicated dicentric r(14) chromosome.  Cells from the patient cultured, harvested, and stained using standard methods for karyotype analysis. A GTG-banded metaphase showing the r(14) chromosome. B The same metaphase was sequentially analyzed by FISH with IGH/BCL2 probes, revealing that the r(14) chromosome lacks the IGH signal. C FISH with NORs probes showed that the r(14) chromosome lacks NORs signals. D FISH with the TCL1 probe indicated that the deleted region is distal to the TCL1 gene. Additionally, two abnormal cells were identified: one with three signals, likely indicating a dicentric r(14), and one with only one signal, suggesting a loss of the r(14) chromosome Microarray studyTo determine the extent of the genomic imbalance, array comparative genomic hybridization (aCGH) was performed using CytoScan™ Technology (Thermo Fisher Scientific Inc). The processes of digestion, ligation, Polymerase chain reaction (PCR), purification of PCR products, quantification, fragmentation, labeling, matrix hybridization, washing, staining, and scanning arrays were carried out following the supplier’s recommendations. Data analysis was conducted with ChAS 4.3 software. Results were interpreted using several databases, including the Database of Genomic Variants, Cytogenomics, Array Group CNV Database, Ensembl Resources, OMIM, UCSC Genome Browser, ClinGen, ClinVar, and CHD wiki. The analysis revealed a terminal deletion of approximately 1.7 Mb on chromosome 14, encompassing 23 genes: BRF1, BTBD6, PACS2, TEX22, MTA1, CRIP2, CRIP1, TEDC1, IGH, TMEM121, LOC105370697, MIR8071-1, MIR8071-2, ELK2, MIR4539, MIR4507, MIR4538, MIR4537, FAM30A, ADAM6, LINC00226, LINC00221 , and MIR5195 (Fig. 2 ). Based on all the studies, the patient’s karyotype was concluded to be 46,XX, r(14).ish r(14)(p11q32.33) (NOR-,TCL1+,IGH-).arr[GRCh38] 14q32.33(105,194,385_106,876,229)x1 dn.  Analysis of aCGH. Left, image representing chromosome 14. Right, plot of weighted log2 ratio. The red arrows indicates the deleted region: arr[GRCh38] 14q32.33 (105,194,385_106,876,229)x1 Ring mitotic instabilityGiven the known mitotic instability of several ring chromosomes, we investigated this phenomenon in our patient. A new culture of the patient’s peripheral blood lymphocytes, stimulated with phytohaemagglutinin, was prepared. Unlike the standard culture used for karyotype analysis, this culture was directly fixed and washed in a cold solution of absolute methanol: glacial acetic acid (3:1) after 72 h of incubation, without exposure to colchicine or hypotonic shock. Cells suspended in fixative solution were dropped onto microscope slides and stained with Giemsa. Cells in metaphase, anaphase, telophase, or cytokinesis were analyzed for mitotic disturbances, and micronucleated cells were counted. Selected microscopic coordinates were recorded for subsequent FISH analyses using IGH and TCL1 break-apart probes. This method revealed various mitotic disturbances. Several cells failed to align at the classical metaphase plate, showing chromosome compaction and distribution similar to those in a classical colchicine block (Fig. 3 A-D). Additionally, some cells displayed chromosome laggards affecting chromosomes other than the r(14) (Fig. 3 E-H) or exhibited a multipolar mitosis-like chromosome organization (Fig. 3 I-L) (for comparison, see Barajas-Torres et al., 2016 [ 10 ]).  Giemsa-stained altered mitotic cells, sequentially studied by FISH using the TCL1 break-apart probe. These cells were harvested without colchicine block and KCl shock. A-D Two metaphase cells with chromosome compaction and distribution resembling those harvested with colchicine. E-H Metaphase cells displaying lagging chromosomes, excluding chromosome 14 and r(14). I-L Mitotic cells with multipolar-like configurations, also showing lagging chromosomes Sequential FISH assays demonstrated that the segregation of the r(14) chromosome was preferentially affected (Fig. 4 : B-C, E-F, and H-I). As a result of this anomalous anaphase-telophase segregation, the r(14) chromosome would be excluded from the main nucleus in the daughter cells, likely forming a micronucleus. The analysis of mitotic figures revealed the following counts of abnormal cells: 33 out of 81 (41%) metaphase cells, 2 out of 10 (20%) anaphase cells, and 4 out of 28 (14%) telophase-cytokinesis cells.  Giemsa-stained mitotic cells serially studied by consecutive FISH with IGH and TCL1 break apart probes (these cells were harvested without both colchicine block and hypotonic shock). A-C Anaphase cell in which a dicentric (duplicated) r(14) is in the middle of the mitotic spindle due probably to the nullification of forces generated by the centromeric traction to opposite poles. D-F Anaphasic r(14) lagging probably caused by a merotelic union. G-I Telophasic cell with r(14) chromosome lagging in both daughter nuclei which, probably, will be a micronucleus in each daughter cell Consistent with these findings, we observed MN in 27 out of 305 (9%) interphase cells. FISH analysis of 21 out of those 27 micronucleated cells confirmed the presence of the r(14) chromosome in all of them (Fig. 5 ).  Selected micronucleated cells studied by consecutive FISH with the IGH and TCL1 break apart probes. A, D, G, J Giemsa-stained micronucleated cells. B, E, H, K Here, micronucleated cells show only one signal of the IGH gene located in the normal chromosome 14. Whereas, in C, F, I, L these cells show two signals of the TCL1 gene; interestingly, one of these signals is in the micronucleus of each cell, thus demonstrating that the micronucleus contains the genetic material of the r(14) chromosome. Furthermore, it is noteworthy that MN in I and L show double TCL1 signal, suggesting the presence of a dicentric (duplicated) ring chromosome Discussion and conclusionsOur patient exhibited typical clinical features of ring chromosome 14 syndrome, including epileptic seizures, craniofacial dysmorphism, and skeletal abnormalities. In 23 reported cases with pure genomic deletions ranging from 0.3 to 5 Mb, including ours (Table 1 ), a consistent phenotype was observed, particularly with epileptic seizures (23/23), intellectual disability (23/23), microcephaly (22/22), speech impairment (22/23), hypotonia (19/21), and facial dysmorphism (17/23). However, other clinical features such as scoliosis (10/18), ocular anomalies (11/20), and susceptibility to infections (10/19) were also present although at a lower frequency. Chromosomal instability in our case led to chromosome 14 monosomy in some cells (12.3%) and duplication of r(14) sequences in others (6.2%) (Fig. 2 -D). This is similar to what has been observed in rings derived from chromosomes other than 14 [ 14 ]. The mechanism behind this co-occurrence involves anaphase segregation after a sister chromatid exchange in the r(14) chromosome [ 2 , 9 ]. However, other mechanisms such as loss of the ring chromosome due to dicentric chromosome formation or merotelic unions may also contribute to chromosome 14 monosomy. Loss of the ring chromosome can also be caused by other mechanisms. When a dicentric duplicated r(14) chromosome is pulled towards opposite poles at anaphase, the traction forces will be nullified. The dicentric chromosome will remain in the middle of the equatorial plaque (as exemplified in Fig. 4 A-C) giving rise to two daughter cells with monosomy, one of which could retain the dicentric chromosome as a micronucleus (as observed in Fig. 5 G-L). Merotelic unions of the r(14) are other mechanisms of origin of monosomy 14; in Fig. 4 D-F an r(14) chromatid is lagged in the middle of the anaphase cell, probably due to a merotelic union, and then, that chromatid will be absent from the nucleus of the daughter cell. A potential third mechanism of r(14) loss could be a delayed alignment in the equatorial plane of the r(14) at metaphase, as suggested in the Fig. 4 G-I, where both r(14) chromatids are lagged during telophase despite an apparent kinetochore-microtubule amphitelic attachment; subsequently, both daughter cells would lose the r(14) chromosome, becoming monosomic. On the other hand, micronucleated cells were detected with a frequency of 9% (27/305). The most significant finding was that all MN from 21 micronucleated cells tested by FISH studies had r(14) chromosome sequences (Fig. 5 ). We have not identified reports of micronucleated cells in patients with an r(14) chromosome. However, evidence from ring chromosomes of other chromosomes indicates a propensity for these rings to be excluded from the main nucleus as MN. Ledbetter et al. [ 15 ] observed in a case of r(15) that 5 out of 1,000 cells were micronucleated and exhibited one or two silver NOR signals, suggesting the presence of monocentric and dicentric r(15) chromosomes in these MN. Similarly, Los et al. [ 16 ] reported 18 chromosome-derived MN in a patient with r(18). Yip et al. [ 17 ] observed MN in 4% of interphase cells and 16% of cells treated with cytochalasin B in a patient with r(3). Using whole chromosome painting (WCP3), they demonstrated chromosome 3 sequences in all MN. Urban et al. [ 18 ] reported 2.8% of cells with MN in a patient with r(6), all of which contained chromosome 6 centromeric sequences. In a case of r(7), Mehraein et al. [ 19 ] observed 3.5% micronucleated cells and confirmed the presence of chromosome 7 material in these MN using FISH with WCP7 and D7Z1 probes. A comparable finding was reported in a case of r(13), with 1% of micronucleated cells and 40% of these MN containing sequences derived from chromosome 13 [ 20 ]. Petter et al. [ 21 ] also found MN in three cases of r(13); all exhibiting chromosome 13 signals, as confirmed by WCP13. These findings support the hypothesis that the structure of ring chromosomes inherently leads to their exclusion from the main nucleus as MN. This hypothesis is further supported by Rudd et al. [ 22 ], who demonstrated experimentally that small ring chromosomes from chromosomes 17 and X missegregate more than normal chromosomes. Additionally, 33 out of 81 (41%) metaphase cells exhibited abnormalities such as disorganized chromosome alignment in the equatorial plane, characterized by lagging chromosomes other than r(14), chromatin over-compaction similar to colchicine blockade, and multipolar spindle configurations (Fig. 3 ). It is unlikely that these abnormalities are solely due to the haploinsufficiency of genes in the deleted region, given that TEDC1 (tubulin epsilon and delta complex 1, required for centriole stability; Breslow et al. [ 23 ]) is the only gene directly related to these findings. A common feature of ring chromosomes, including r(14), is the absence or reduction of the canonical telomeric sequence TTAGGG. Studies on telomere dynamics in interphase lymphocytes show that telomeres are located near the central nuclear region, whereas during postmitotic assembly, they move to the nuclear periphery [ 24 , 25 ]. Interestingly, during the G2 phase, telomeres assemble into a disk structure [ 26 , 27 ]. The impact of this telomere positioning on mitosis has not been studied. If the telomeric disk observed in G2 persisted into mitosis, it could act as an anchor for microtubule-kinetochore attachment, ensuring proper chromosome orientation in the equatorial plane and preventing monotelic, syntelic, and merotelic attachments [ 28 ]. A delay in microtubule-kinetochore binding or chromosome misorientation due to the lack of telomeric sequences on ring chromosomes could activate the spindle attachment checkpoint (SAC) [ 29 , 30 , 31 ], arresting metaphase progression until the issue is resolved. It has been noted that a single unattached kinetochore can activate the SAC and inhibit mitotic progression [ 32 ]. A prolonged prometaphase arrest triggers cellular responses such as apoptosis, DNA damage repair activation [ 33 , 34 ], telomere instability [ 35 ], and affects daughter cell proliferation [ 36 ]. Thus, SAC activation could explain the observed instability in many ring chromosome cases, including our r(14) case (Fig. 3 ), characterized by misaligned chromosomes, over-compacted chromosomes, and multipolar spindle configurations, which disrupt centriole duplication independently of cell-cycle progression [ 37 , 38 , 39 ]. The coalescence of extra centrosomes might explain findings by Rivera and Dominguez [ 40 ], who observed hypodiploidy (34 to 45 chromosomes) and polyploidy (74 to 96 chromosomes) in a patient with a r(4) chromosome. In conclusion, ring chromosome syndrome is a pathological entity with significant variability due to factors including the affected chromosome, gene loss, positional effects of genes near breakpoints, and unbalanced segregation following ring chromosome sister chromatid exchange. Although previous work has suggested instability associated with chromosome rings, the evidence has been inconclusive [ 15 , 16 , 17 , 18 , 19 , 20 , 21 ]. Remarkably, we have demonstrated an instability of the r(14) chromosome, mainly involving anaphasic lags and its exclusion from the nucleus in the form of a micronucleus. Further research into the inherent instability of ring chromosomes is needed to clarify these observations in patients with ring chromosomes. Data availabilityNo datasets were generated or analysed during the current study. AbbreviationsArray comparative genomic hybridization Fluorescense in situ hybridization G-bands by trypsin using Giemsa Micronuclei Nucleolus organizer region Online Mendelian inheritance in man Polymerase chain reaction Spindle attachment checkpoint Rinaldi B, Vaisfeld A, Amarri S, Baldo C, Gobbi G, Magini P, et al. Guideline recommendations for diagnosis and clinical management of Ring 14 syndrome-first report of an ad hoc task force. Orphanet J Rare Dis. 2017;12:69. Article PubMed PubMed Central Google Scholar Zollino M, Ponzi E, Gobbi G, Neri G. The ring 14 syndrome. Eur J Med Genet. 2012;55:374–80. Article PubMed Google Scholar Ivanoff AE, Ivanoff CS. Ring chromosome 14 syndrome: what the dentist should know to manage children with r(14) effectively. Folia Med (Plovdiv). 2023;28:65:20–9. Article Google Scholar Giovannini S, Marangio L, Fusco C, Scarano A, Frattini D, Della Giustina E, et al. Epilepsy in ring 14 syndrome: a clinical and EEG study of 22 patients. Epilepsia. 2013;54:2204–13. Zampini L, Zanchi P, D’Odorico L. Developing with ring 14 syndrome: a survey in different countries. Clin Linguist Phon. 2014;28:844–56. Zollino M, Seminara L, Orteschi D, Gobbi G, Giovannini S, Giustina ED, et al. The ring 14 syndrome: clinical and molecular definition. Am J Med Genet Part A. 2009;149A:1116–24. Article CAS PubMed Google Scholar Nikitina TV, Kashevarova AA, Gridina MM, Lopatkina ME, Khabarova AA, Yakovleva YS, et al. Complex biology of constitutional ring chromosomes structure and (in)stability revealed by somatic cell reprogramming. Sci Rep. 2021;11:4325. Article CAS PubMed PubMed Central Google Scholar Sodré CP, Guilherme RS, Meloni VF, Brunoni D, Juliano Y, Andrade JA, et al. Ring chromosome instability evaluation in six patients with autosomal rings. Genet Mol Res. 2010;9:134–43. Guilherme RS, Meloni VF, Kim CA, Pellegrino R, Takeno SS, Spinner NB, et al. Mechanisms of ring chromosome formation, ring instability and clinical consequences. BMC Med Genet. 2011;12:171. Barajas Torres RL, Domínguez Cruz MD, Borjas Gutiérrez C, Ramírez Dueñas ML, Magaña Torres MT. González García JR. 1,2:3,4-Diepoxybutane induces multipolar mitosis in cultured human lymphocytes. Cytogenet Genome Res. 2016;148:179–84. Chen C, Wu LW, He F, Yang LF, Miao P, Ma YP, Wang XL, Peng J. [Ring 14 chromosome syndrome in a boy mainly manifesting as drug-resistant epilepsy]. Zhongguo Dang Dai Er Ke Za Zhi. 2017;19:949–51. PubMed Google Scholar Salter CG, Baralle D, Collinson MN, Self JE. Expanding the ocular phenotype of 14q terminal deletions: a novel presentation of microphthalmia and coloboma in ring 14 syndrome with associated 14q32.31 deletion and review of the literature. Am J Med Genet Part A. 2016;170A:1017–22. Zampini L, Zanchi P, Rinaldi B, Novara F, Zuffardi O. Developmental trends of communicative skills in children with chromosome 14 aberrations. Eur J Pediatr. 2017;176:455–64. Hu Q, Chai H, Shu W, Li P. Human ring chromosome registry for cases in the Chinese population: re-emphasizing cytogenomic and clinical heterogeneity and reviewing diagnostic and treatment strategies. Mol Cytogenet. 2018;11:19. Ledbetter DH, Riccardi VM, Au WW, Wilson DP, Holmquist GP. Ring chromosome 15: phenotype, Ag-NOR analysis, secondary aneuploidy, and associated chromosome instability. Cytogenet Cell Genet. 1980;27:111–22. Los FJ, van den Berg C, Braat PG, Cha’ban FK, Kros JM, Van Opstal D. Ring chromosome 18 in a fetus with only facial anomalies. Am J Med Genet. 1996;66:216–20. Yip MY, MacKenzie H, Kovacic A, McIntosh A. Chromosome 3p23 break with ring formation and translocation of displaced 3p23–>pter segment to 6pter. J Med Genet. 1996;33:789–92. Urban M, Bommer C, Tennstedt C, Lehmann K, Thiel G, Wegner RD, et al. Ring chromosome 6 in three fetuses: case reports, literature review, and implications for prenatal diagnosis. Am J Med Genet. 2002;108:97–104. Mehraein Y, Ehlhardt S, Wagner A, Göttert E, Tilgen W, Zang KD, et al. Somatic mosaicism of chromosome 7 in a highly proliferating melanocytic congenital naevus in a ring chromosome 7 patient. Am J Med Genet A. 2004;131:179–85. Amor DJ, Voullaire L, Bentley K, Savarirayan R, Choo KH. Mosaic monosomy of a neocentric ring chromosome maps brachyphalangy and growth hormone deficiency to 13q31.1-13q32.3. Am J Med Genet A. 2005;133:151–7. Petter C, Moreira LMA, Riegel M. Towards new approaches to evaluate dynamic mosaicism in ring chromosome 13 syndrome. Case Rep Genet. 2019;2019:7250838. PubMed PubMed Central Google Scholar Rudd MK, Mays RW, Schwartz S, Willard HF. Human artificial chromosomes with alpha satellite-based de novo centromeres show increased frequency of nondisjunction and anaphase lag. Mol Cell Biol. 2003;23:7689–97. Breslow DK, Hoogendoorn S, Kopp AR, Morgens DW, Vu BK, Kennedy MC, et al. A CRISPR-based screen for hedgehog signaling provides insights into ciliary function and ciliopathies. Nat Genet. 2018;50:460–71. Amrichová J, Lukásová E, Kozubek S, Kozubek M. Nuclear and territorial topography of chromosome telomeres in human lymphocytes. Exp Cell Res. 2003;289:11–26. Crabbe L, Cesare AJ, Kasuboski JM, Fitzpatrick JA, Karlseder J. Human telomeres are tethered to the nuclear envelope during postmitotic nuclear assembly. Cell Rep. 2012;2:1521–9. Chuang TC, Moshir S, Garini Y, Chuang AY, Young IT, Vermolen B, et al. The three-dimensional organization of telomeres in the nucleus of mammalian cells. BMC Biol. 2004;2:12. Vermolen BJ, Garini Y, Mai S, Mougey V, Fest T, Chuang TC, et al. Characterizing the three-dimensional organization of telomeres. Cytometry A. 2005;67:144–50. Manic G, Corradi F, Sistigu A, Siteni S, Vitale I. Molecular regulation of the spindle assembly checkpoint by kinases and phosphatases. Int Rev Cell Mol Biol. 2017;328:105–61. Rhind N, Russell P. Signaling pathways that regulate cell division. Cold Spring Harb Perspect Biol. 2012;4:a005942. Foley EA, Kapoor TM. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol. 2013;14:25–37. Tan CH, Gasic I, Huber-Reggi SP, Dudka D, Barisic M, Maiato H, et al. The equatorial position of the metaphase plate ensures symmetric cell divisions. Elife. 2015;4:e05124. Rieder CL, Cole RW, Khodjakov A, Sluder G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol. 1995;130:941–8. Orth JD, Loewer A, Lahav G, Mitchison TJ. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol Biol Cell. 2012;23:567–76. Colin DJ, Hain KO, Allan LA, Clarke PR. Cellular responses to a prolonged delay in mitosis are determined by a DNA damage response controlled by Bcl-2 family proteins. Open Biol. 2015;5:140156. Hayashi MT, Cesare AJ, Fitzpatrick JA, Lazzerini-Denchi E, Karlseder J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol. 2012;19:387–94. Uetake Y, Sluder G. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol. 2010;20:1666–71. Pihan GA. Centrosome dysfunction contributes to chromosome instability, chromoanagenesis, and genome reprograming in cancer. Front Oncol. 2013;3:277. Maiato H, Logarinho E. Mitotic spindle multipolarity without centrosome amplification. Nat Cell Biol. 2014;16:386–94. Aydogan MG, Steinacker TL, Mofatteh M, Wilmott ZM, Zhou FY, Gartenmann L, et al. An autonomous oscillation times and executes centriole biogenesis. Cell. 2020;181:1566–81. Rivera H, Domínguez MG. Variegated aneuploidy and ring chromosome syndromes overlap. Am J Med Genet A. 2010;152:228–9. Download references AcknowledgementsWe thank patient’s parents for their cooperation in the conducted studies. Not applicable. Author informationAuthors and affiliations. Facultad de Medicina Matamoros, Universidad Autónoma de Tamaulipas, Matamoros, Tamps, México Juan Pablo Meza-Espinoza División de Genética, Centro de Investigación Biomédica de Occidente, Instituto Mexicano del Seguro Social (IMSS), Guadalajara, Jalisco, México Juan Ramón González-García Maestría en Ciencias en Biomedicina Molecular, Facultad de Medicina, Universidad Autónoma de Sinaloa, Culiacán, Sin, México Nayeli Nieto-Marín & Liliana Itzel Patrón-Baro Doctorado en Genética Humana, Universidad de Guadalajara, Guadalajara, Jalisco, México Rosa María González-Arreola Facultad de Ciencias Químico-Biológicas, Universidad Autónoma de Sinaloa, Culiacán, Sin, México Eliakym Arámbula-Meraz Facultad de Odontología, Universidad Autónoma de Sinaloa, Culiacán, Sin, México Julio Benítez-Pascual Facultad de Medicina, Universidad Autónoma de Sinaloa, Culiacán, Sin, México Alberto Kousuke De la Herrán-Arita, Marco Antonio Valdez-Flores, Tomás Adrián Carrillo-Cázares & Verónica Judith Picos-Cárdenas Facultad de Biología, Universidad Autónoma de Sinaloa, Culiacán, Sin, México Claudia Desireé Norzagaray-Valenzuela You can also search for this author in PubMed Google Scholar ContributionsJ.P.M.E. Data collection and article drafting. J.R.G.G. and R.M.G.A. Performing and interpreting the cytogenetic studies and providing intellectual input during the manuscript drafting. N.N.M., L.I.P.B., and E.A.M. Collection of samples, making, and interpretation microarrays studies. J.B.P., M.A.V.F., C.D.N.V., and T.A.C.C. Patient recruitment and clinical examination. A.K.D.A. Intellectual input during discussion and revision of the draft. V.J.P.C. Conception and design of the study, drafting, acquisition and interpretation of data. All the authors contributed to the critical review and approved the final version of the manuscript. Corresponding authorCorrespondence to Verónica Judith Picos-Cárdenas . Ethics declarationsEthics approval and consent to participate. This study was approved by the ethics committees of all participating institutions. Consent for publicationThe patient’s parents provided informed consent for the use of biological material for research purposes, but they objected to publish photographs. Competing interestsThe authors declare no competing interests. Additional informationPublisher’s note. Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Rights and permissionsOpen Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data. Reprints and permissions About this articleCite this article. Meza-Espinoza, J.P., González-García, J.R., Nieto-Marín, N. et al. Chromosomal instability in a patient with ring chromosome 14 syndrome: a case report. Mol Cytogenet 17 , 17 (2024). https://doi.org/10.1186/s13039-024-00686-0 Download citation Received : 25 May 2024 Accepted : 26 June 2024 Published : 18 July 2024 DOI : https://doi.org/10.1186/s13039-024-00686-0 Share this articleAnyone you share the following link with will be able to read this content: Sorry, a shareable link is not currently available for this article. Provided by the Springer Nature SharedIt content-sharing initiative
Molecular CytogeneticsISSN: 1755-8166
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
IMAGES
VIDEO
COMMENTS
The diagnosis is often delayed because Cushing's is frequently masked by its overlap with more common medical problems such as diabetes, high blood pressure, obesity, and polycystic ovary syndrome. Cushing's may be more common than previously thought. In this case, a 32-year-old female was ill for at least one and a half years before diagnosis.
A population-based study in Denmark reported the incidence of adrenal adenoma as 0.6 and adrenal carcinoma as 0.2 per million per year . In 1.3 to 8.7 percent of adults AT are discovered incidentally. ... followed by treatment for CS as we did in case 5, while severe Cushing's syndrome, when diagnosed in the 1st trimester of pregnancy, ...
In one series, Cushing's syndrome occurred in 3 of 16 patients with the multiple endocrine neoplasia syndrome and the Zollinger-Ellison syndrome (16 percent). 8 In these cases, mild Cushing's ...
Cushing's disease is a form of Cushing's syndrome with overproduction of adrenocorticotropic hormone (ACTH) due to pituitary adenoma. The diagnosis is made using clinical features and paraclinical tests including urinary free cortisol (UFC), serum ACTH, dexamethasone suppression tests (DSTs), pituitary magnetic resonance imaging (MRI), and sometimes by inferior petrosal sinus sampling ...
An interactive case study on the differential diagnosis of Cushing's syndrome. This module looks at the diagnostic strategy required to establish the cause of adrenocorticotrophic hormone (ACTH)-dependent Cushing's syndrome, when imaging and biochemical results are inconclusive. Go to case study. Posted on October 11, 2022.
Background Cushing's syndrome occurs due to overproduction of cortisol from adrenal glands. Endogenous hypercortisolemia can occur secondary to adrenocorticotropic hormone (ACTH) dependent as well as independent causes. The presence of non-specific symptoms and signs contributes to a delay in diagnosis. Early identification and prompt definitive management is crucial. It is important to be ...
In most case series of Cushing's syndrome, an average of 8% of patients have adrenal carcinoma. 21 If 6 per million is 8% of the group, the total Cushing's syndrome group is 75 persons per ...
Cushing syndrome is a rare disease that rarely presents as acute psychosis. In this case, the patient presented with acute psychosis and agitation as the first manifestations of the disease which led to the admission of the patient to a psychiatry hospital for one month, as it was difficult to restrain her sufficiently for performing appropriate diagnostic tests due to disturbing behavior.
Purpose: To present a case study of a 34-year-old woman with Cushing's disease and provide nurse practitioners (NPs) with the understanding of the clinical presentation needed for early recognition and treatment of the disease. Data sources: A comprehensive review of published literature on Cushing's disease. Findings from history, physical examination, and diagnostic studies of a woman ...
Introduction: Cushing's syndrome is caused by an extended exposure to increased levels of endogenous or exogenous glucocorticoids. It is a syndrome that can be extremely challenging to diagnose as many symptoms and signs are also indications of other disease processes. ... Case: A 76 year old man presented to hospital with a six month history ...
Case 144 -- Cushing's Syndrome. Contributed by Sanja Dacic, MD and Prabha B. Rajan, MD Published on line in April 1998. The patient is a 41-year-old Caucasian female who was admitted to the hospital for evaluation of high blood cortisol level. Her complaints were fatigue, weakness, lethargy, decreased concentration and decreased memory over the ...
Check out this nursing case study on cushing syndrome & learn everything you will need to about to ace your NCLEX questions. View the lesson! Cushing's Syndrome Case Study (60 min) ... Each case study was written by experienced nurses with first hand knowledge of the "real-world" disease process.
Abstract. Cushing's syndrome (CS) is considered a rare disease. The most common cause is the exogenous use of glucocorticoids (GCs), which are often given within a controlled medical setting, but their factitious use is rare. Factitious CS is more common in females, young patients, those with psychiatric disorders, and those with contacts ...
A probabilistic model for Cushing's syndrome screening in at-risk populations: a prospective multicenter study. J Clin Endocrinol Metab. 2016;101(10):3747-3754. doi pubmed; Lindholm J, Juul S, Jorgensen JO, Astrup J, Bjerre P, Feldt-Rasmussen U, Hagen C, et al. Incidence and late prognosis of cushing's syndrome: a population-based study.
Moreover, another group reported the case of a patient with Cushing's disease followed by ectopic Cushing's syndrome more than 30 years later (8). To our knowledge, however, we here describe the first case report on a single patient with a cortisol-producing adrenocortical adenoma and subsequent CD.
We present a case report with clinical, laboratory, and imaging data and discuss the literature pertaining to pregnancy in patients with Cushing's syndrome. ... An unstimulated inferior petrosal sinus sampling study showed a gradient to the right (ACTH on the right was 1,007 and 927 pg/mL, on the left was 408 and 216 pg/mL, and in the ...
The study analyzed death data from more than 19,000 patients in 92 studies published through January 2021. "Our results found that death rates have fallen since 2000 but are still unacceptably high," Limumpornpetch said. Cushing's syndrome affects many parts of the body because cortisol responds to stress, maintains blood pressure and ...
The third case was a 25-year-old Iranian man who presented with symptoms and signs of Cushing's syndrome. Pituitary magnetic resonance imaging revealed a microadenoma 5 × 9 mm. Whole-body scan showed abnormal focal somatostatin receptors analog avid lesion in the posterior aspect of inferior third of right lung, highly suggestive of ectopic ...
Definition. Cushing syndrome is an endocrine disease with multiple etiologies and is characterized by a constellation of clinical manifestations that result from excessive concentrations of circulat-ing cortisol (i.e., hypercortisolism). The syndrome is named after Harvey Cushing, an American surgeon who first reported the condition in 1912.
The urinary free cortisol (UFC) test is widely used for the screening of Cushing's syndrome. This case study illustrates the potential failure of the UFC test to correctly diagnose Cushing's disease (CD), indicating that the use of other complementary tests may be necessary to diagnose this disease in some cases.
Abstract. Cushing's syndrome is a rare endocrine disorder that comprises a large group of signs and symptoms resulting from chronic exposure to excess corticosteroids. Most cases of Cushing's syndrome are due to increased adrenocorticotropic hormone production from a pituitary adenoma, which is referred to as Cushing's disease. Most of the signs and symptoms are nonspecific and common in ...
Purpose Early diagnosis and immediate treatment of Cushing's syndrome (CS) are critical for a better prognosis but remain a challenge. However, few comprehensive reports have focused on this issue or investigated whether patient-reported manifestations are consistent with physician-assessed symptoms of CS. This study aimed to clarify the differences in patient-reported and physician-assessed ...
Glucocorticoid withdrawal syndrome can be difficult to differentiate from adrenal insufficiency, which complicates glucocorticoid dosing, and tapering regimens. In a retrospective study of the postoperative course of 81 patients with adrenocorticotropic hormone independent Cushing syndrome, glucocorticoid withdrawal syndrome was most common ...
Cushing's syndrome (CS) during pregnancy is a rare endocrine disorder characterized by hypercortisolism, which is significantly associated with maternal-fetal complications. Despite its rarity, CS during pregnancy may be related to a high risk of complications for both the mother and fetus.The aim of the present case study is to update the ...
This case evolved into Cushing's syndrome due to ectopic ACTH secretion syndrome (EAS) provoked by liver metastasis originating from a pancreatic neuroendocrine carcinoma (NEC), an occurrence not previously documented in MEN1 literature for pancreatic NECs. ... This study was approved by the Institutional Review Board of the Instituto ...
Stavres, J., Vallecillo-Bustos, A., Newsome, T.A. et al. Hemodynamic responses to the cold pressor test in individuals with metabolic syndrome: a case-control study in a multiracial sample of adults.
Background To evaluate the efficacy of SIMBA as an educational intervention for both HCPs and people with either PCOS or adrenal conditions and to study the change in knowledge of people with PCOS or adrenal conditions about the conditions and expectations from the HCPs involved in their care following SIMBA-PPI sessions. Methods Two SIMBA-PPI sessions (SIMBA-PPI Polycystic ovary syndrome ...
Background Ring chromosome 14 syndrome is a rare disorder primarily marked by early-onset epilepsy, microcephaly, distinctive craniofacial features, hypotonia, intellectual disability, and delay in both development and language acquisition. Case presentation A 21-year-old woman with a history of epileptic seizures since the age of 1.5 years presented with distinctive craniofacial features ...